
Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas

 ,
, 
 , , ,
, , ,  ,
,  ,
, 
 , , , ,
, , , ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structures and 3D Modeling
2.2. Protein–Protein Docking
3. Results
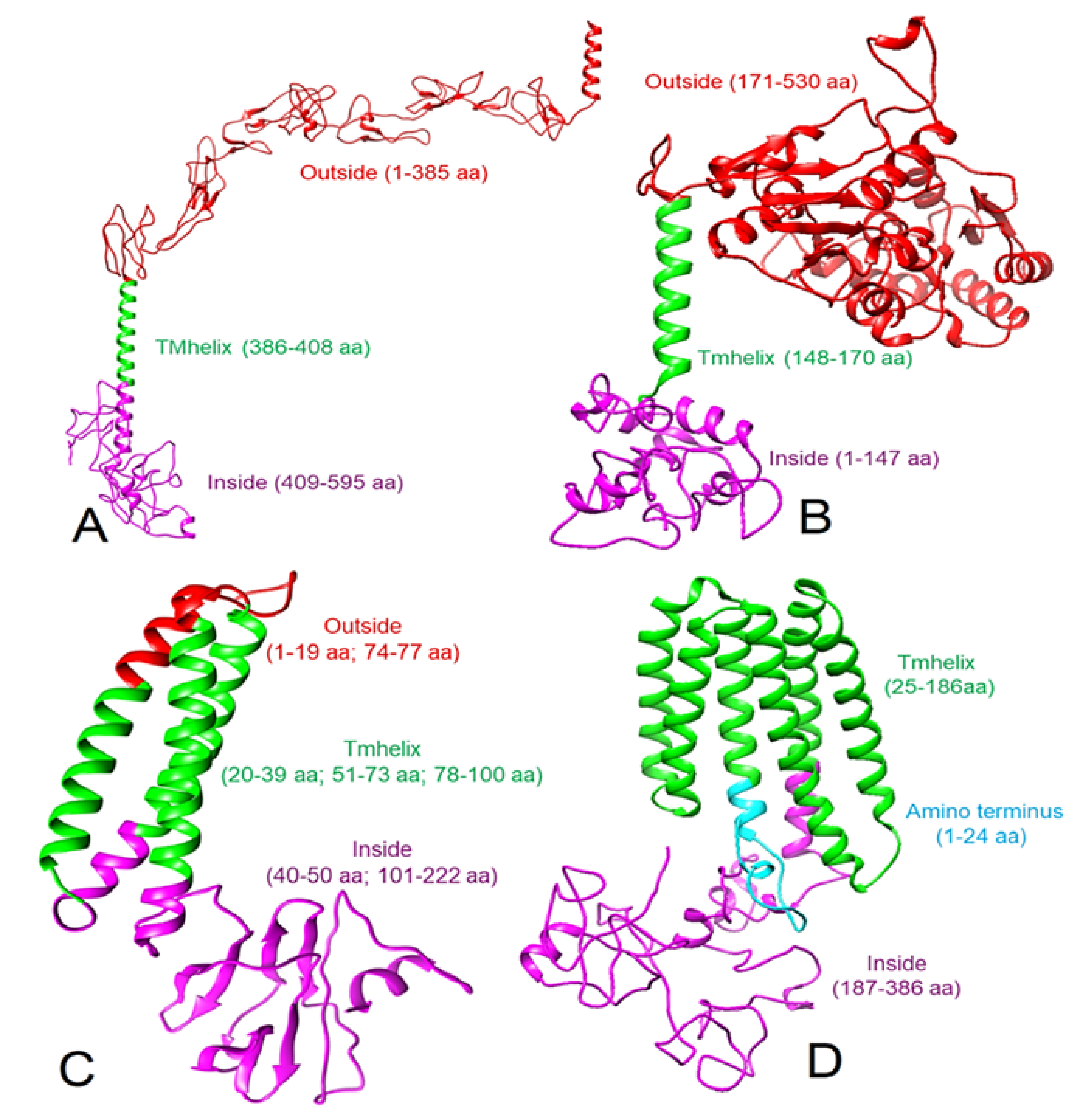
3.1. 3D Protein Models
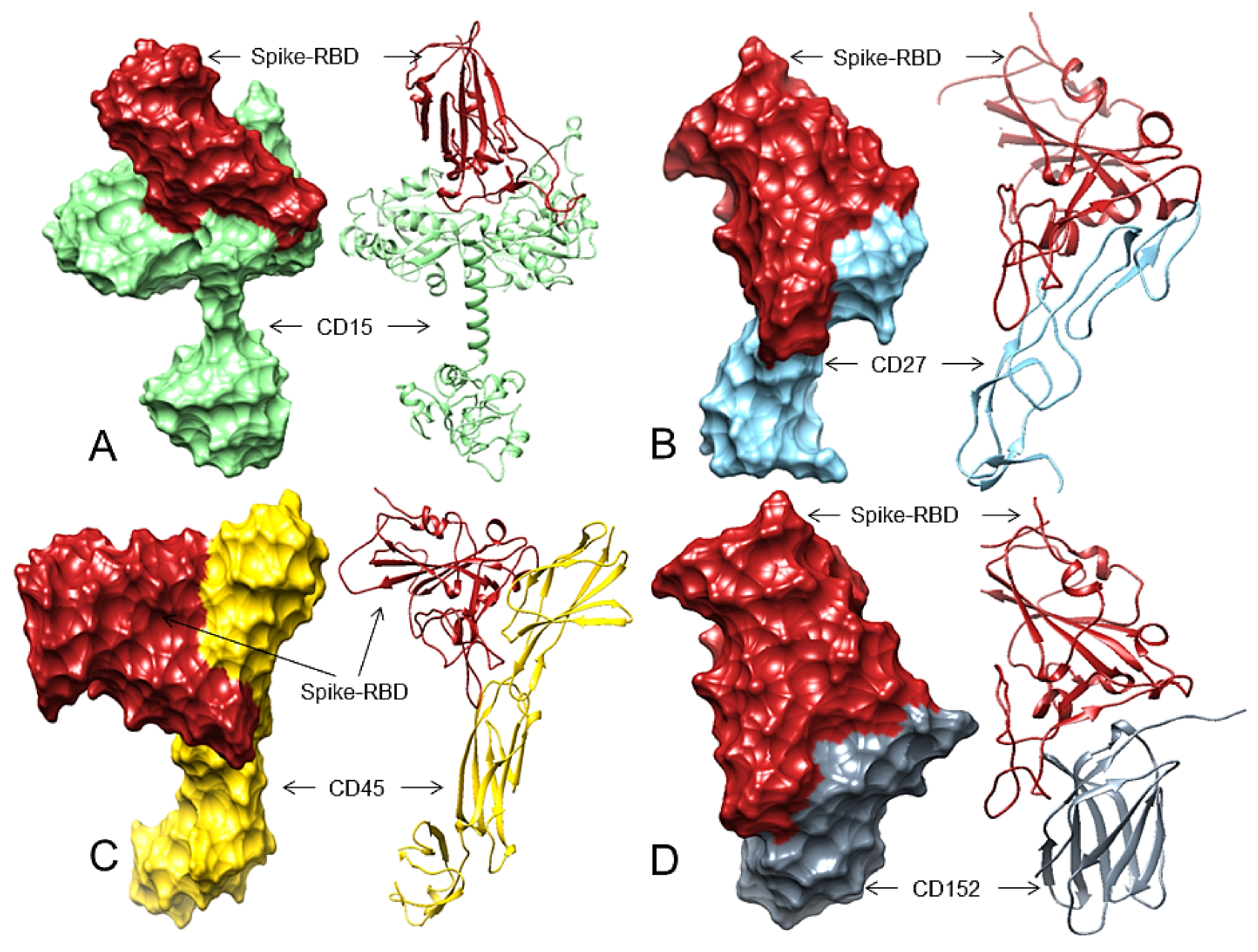
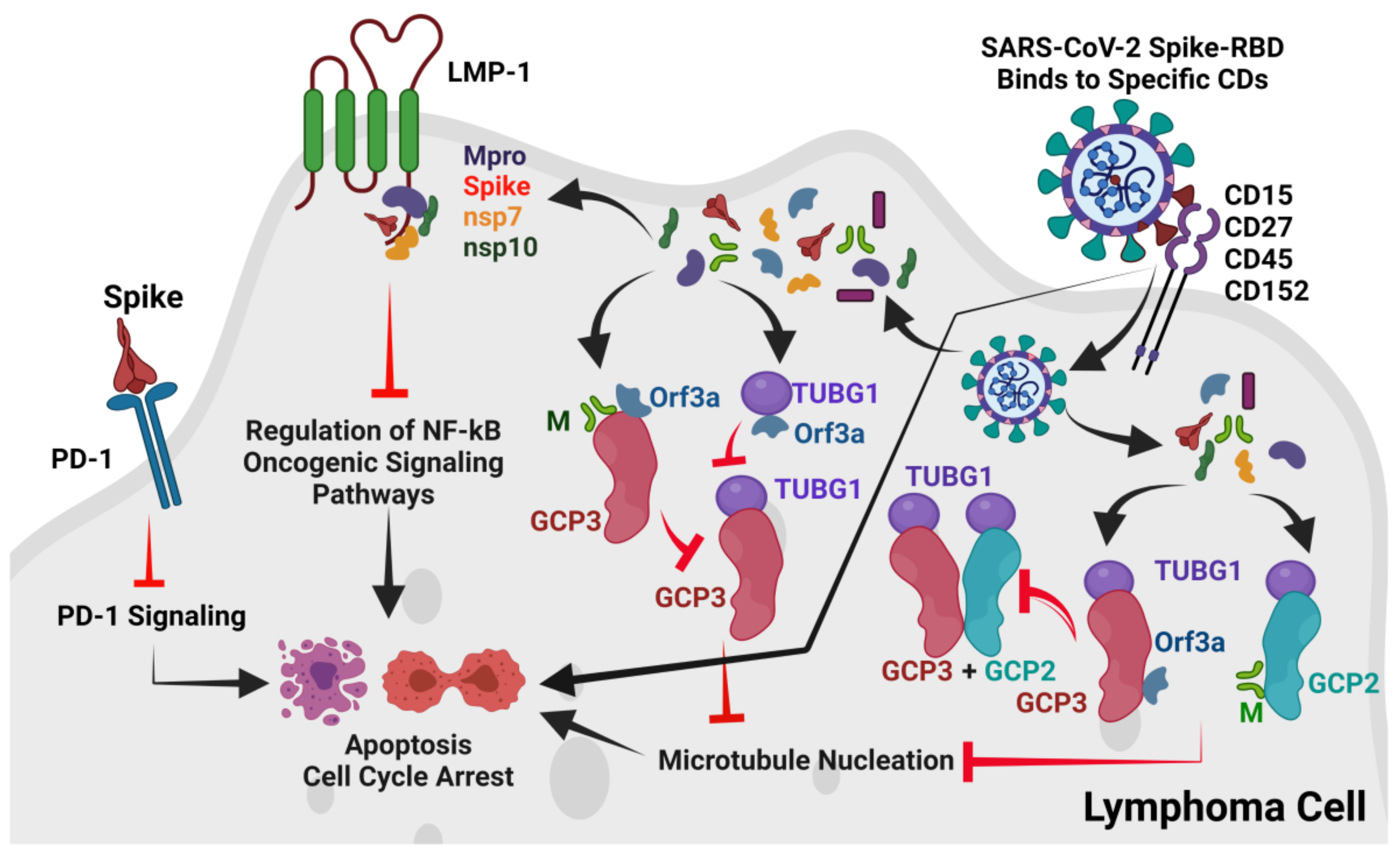
3.2. Spike-RBD May Bind to CD15, CD27, CD45, and CD152 Receptors of cHL or FL
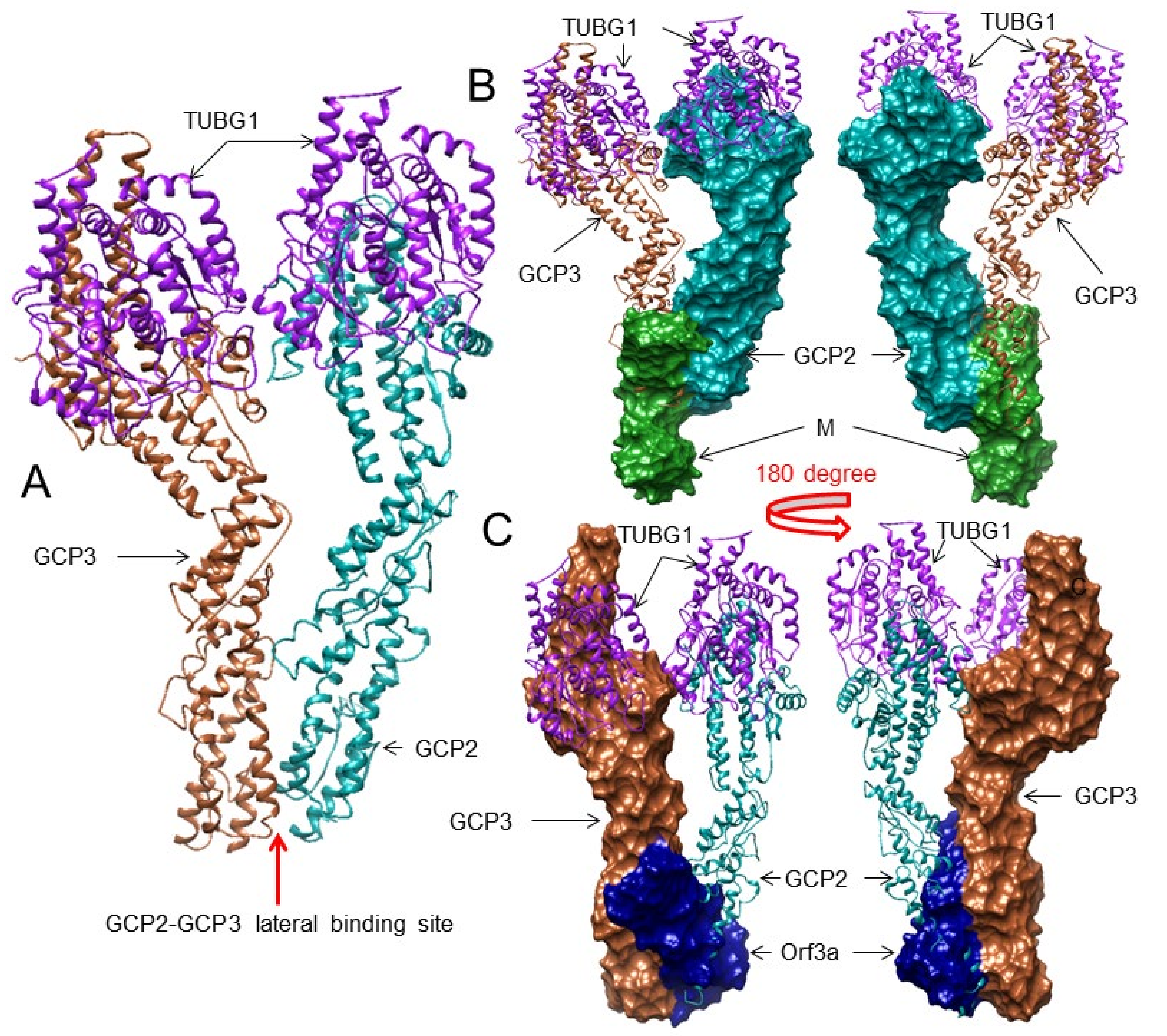
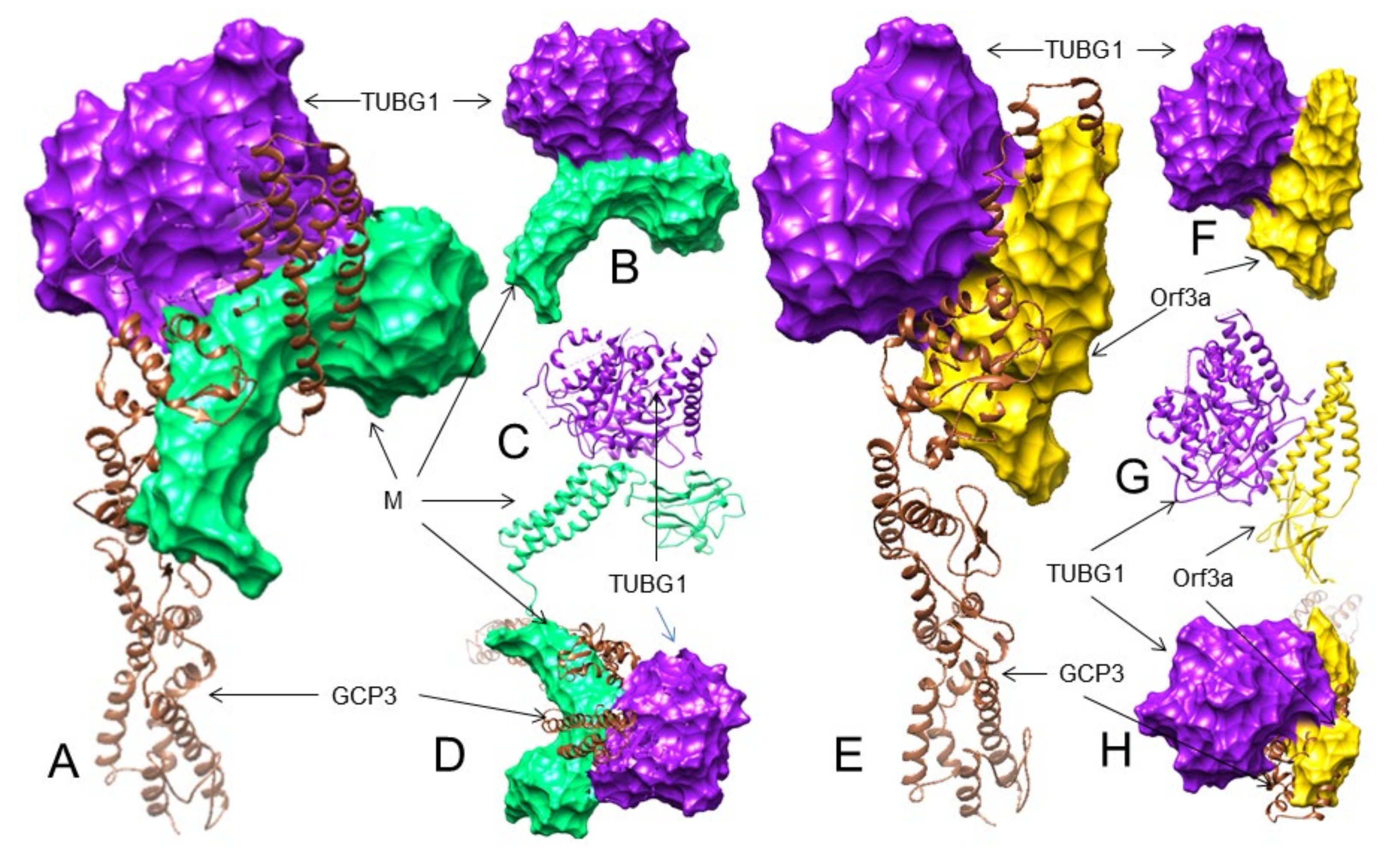
3.3. The SARS-CoV-2 M and ORF3a Proteins Interact with the Human Gamma-Tubulin Complex Components and May Inhibit Tubulin Nucleation
3.4. The M and ORF3a Proteins May Interact at GCP2–GCP3 Lateral Binding Sites
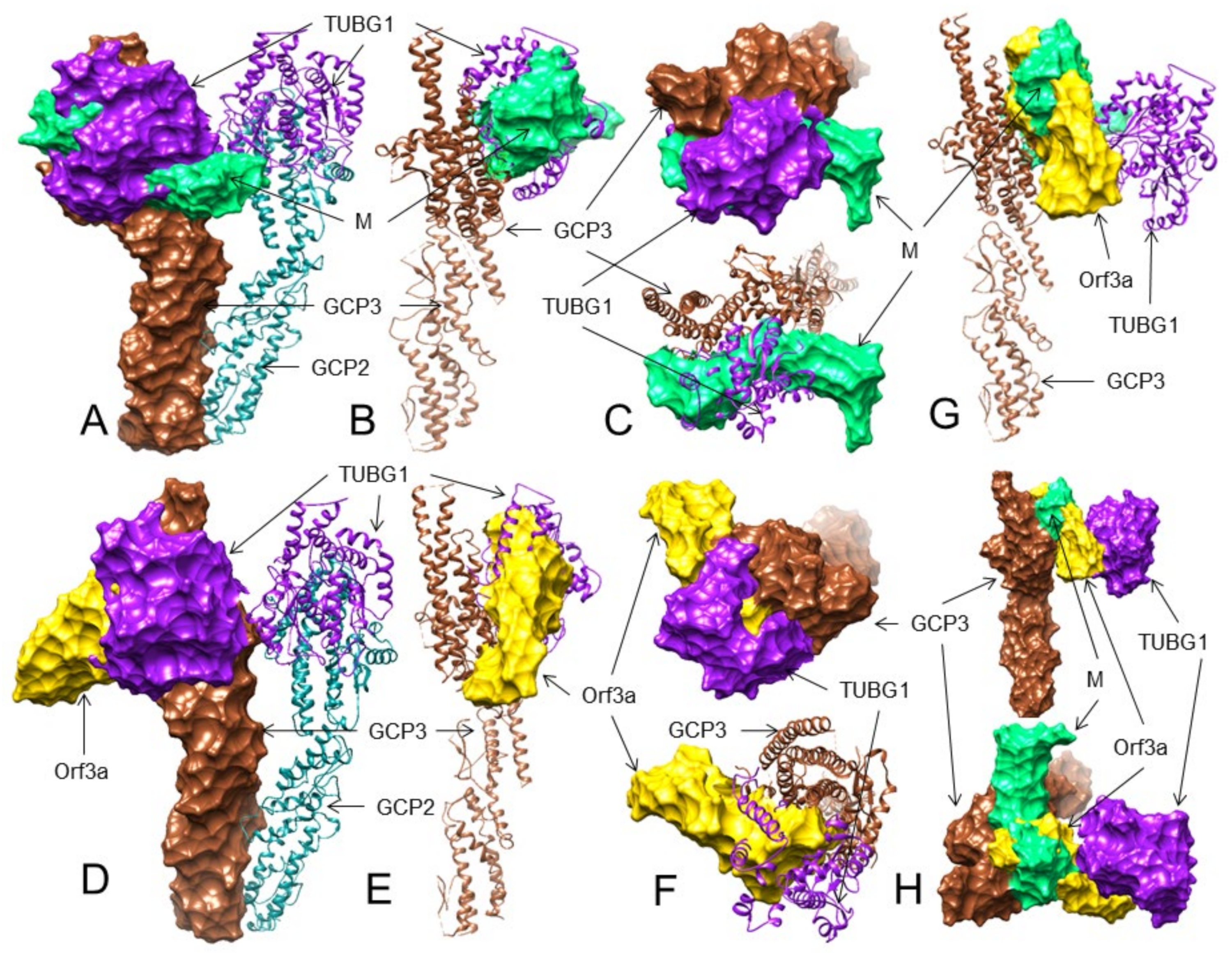
3.5. Both the M and ORF3a Proteins May Interact with GCP3 at Its TUBG1 Binding Sites
3.6. The M but Not the ORF3a Protein May Strongly Interact with TUBG1 at Its GCP3 Binding Sites
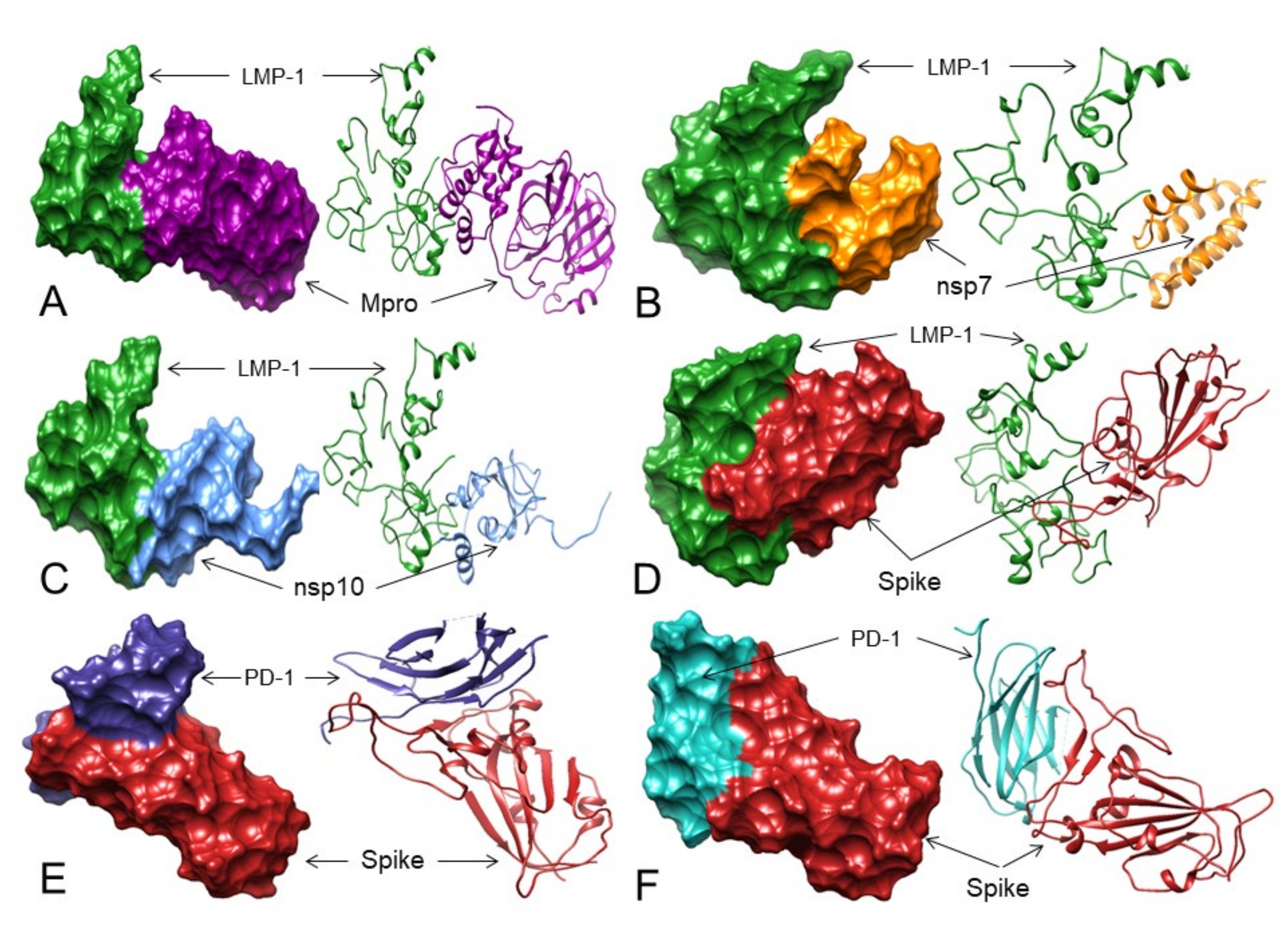
3.7. LMP-1 Signaling Domains of EBV Potentially Interact with SARS-CoV-2 Proteins
3.8. Does the Spike Protein Interact with PD-1 to Block Access to PD-L1 and PD-L2?
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| hACE2 | Human Angiotensin-converting enzyme 2 |
| BSAP | B-cell-specific transcription factor |
| CD | Cluster of differentiation |
| CD15 | CD antigen 15 (alpha-(1,3)-fucosyltransferase 4, FUT4) |
| CD20 | CD antigen 20 (B-lymphocyte antigen CD20) |
| CD27 | CD antigen 27 (Tumor necrosis factor receptor superfamily member 7, TNFRSF7) |
| CD30 | CD antigen 30 (tumor necrosis factor receptor superfamily member 8, TNFRSF8) |
| CD40 | CD antigen 40 (Tumor necrosis factor receptor superfamily member 5, TNFRSF5) |
| CD45 | CD antigen 45 (Receptor-type tyrosine-protein phosphatase C, PTPRC) |
| CD80 | CD antigen 80 (T-lymphocyte activation antigen CD80) |
| CD86 | CD antigen 86 (T-lymphocyte activation antigen CD86) |
| CD95 | CD antigen 95 (Tumor necrosis factor receptor superfamily member 6, TNFRSF6, or Apoptosis-mediating surface antigen FAS) |
| CD125 | CD antigen 125 (Cytotoxic T-lymphocyte protein 4, CTLA4) |
| cHL | Classical Hodgkin lymphoma |
| COVID-19 | Coronavirus Disease 2019 |
| CTLA4 | T-lymphocyte associated protein 4 (CD antigen 125, CD125) |
| DLBCL | Diffuse large B-cell lymphoma |
| EBV | Epstein–Barr virus |
| FL | Follicular lymphoma |
| FUT4 | Alpha-(1,3)-fucosyltransferase 4 (CD antigen 15, CD15) |
| GCP2 | Gamma-tubulin complex component 2 |
| GCP3 | Gamma-tubulin complex component 3 |
| GGT1 | Gamma-glutamyl transferase 1 |
| HRS | Hodgkin or Reed–Sternberg cells |
| IRF4 | Interferon regulatory factor 4 |
| JAK | Janus kinase 1 |
| mABs | Monoclonal antibodies |
| LMP-1 | Latent oncogenic membrane protein 1 |
| M | Membrane protein of SARS-CoV-2 |
| MMAE | Monomethyl auristatin E |
| MS4A1 | Membrane-spanning 4-domains subfamily A member 1 |
| MT | Microtubule |
| PAX5 | Paired box protein Pax-5 |
| MUM1 | Multiple myeloma oncogene 1 |
| PD-1 | Programmed cell death-1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| PD-L2 | Programmed cell death 1 ligand 2 |
| PTPRC | Receptor-type tyrosine-protein phosphatase C (CD antigen 45, CD45) |
| SARS-CoV-2 | Severe acute respiratory coronavirus 2 |
| RBD | Receptor binding domain of spike protein |
| S | Spike protein of SARS-CoV-2 |
| STAT | Signal transducer and activator of transcription |
| TNFRSF5 | Tumor necrosis factor receptor superfamily member 5 (CD antigen 40, CD40) |
| TNFRSF6 | Tumor necrosis factor receptor superfamily member 6 (CD antigen 95, CD95) |
| TNFRSF7 | Tumor necrosis factor receptor superfamily member 7 (CD antigen 27, CD27) |
| TNFRSF8 | Tumor necrosis factor receptor superfamily member 8 (CD antigen 30) |
| TRAFs | Tumor necrosis factor receptor-associated factors |
| TRADD | Tumor necrosis factor receptor type 1-associated DEATH domain protein |
| TUBG1 | Tubulin gamma-1 chain |
References
- Jarahzadeh, M.H.; Asadian, F.; Farbod, M.; Meibodi, B.; Abbasi, H.; Jafari, M.; Raee-Ezzabadi, A.; Bahrami, R.; Neamatzadeh, H. Cancer and Coronavirus Disease (COVID-19): Comorbidity, Mechanical Ventilation, and Death Risk. J. Gastrointest. Cancer 2021, 52, 80–84. [Google Scholar] [CrossRef]
- Challenor, S.; Tucker, D. SARS-CoV-2-induced remission of Hodgkin lymphoma. Br. J. Haematol. 2021, 192, 415. [Google Scholar] [CrossRef] [PubMed]
- Sollini, M.; Gelardi, F.; Carlo-Stella, C.; Chiti, A. Complete remission of follicular lymphoma after SARS-CoV-2 infection: From the “flare phenomenon” to the “abscopal effect”. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2652–2654. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef] [PubMed]
- Chioccam, E.A. Oncolytic Viruses. Nat. Rev. 2002, 2, 938–950. [Google Scholar] [CrossRef]
- Kurokawa, C.; Iankov, I.D.; Anderson, S.K.; Aderca, I.; Leontovich, A.A.; Maurer, M.J.; Oberg, A.L.; Schroeder, M.A.; Giannini, C.; Greiner, S.M.; et al. Constitutive Interferon Pathway Activation in Tumors as an Effiacy Determinant Following Oncolytic Virotherapy. J. Natl. Cancer Inst. 2018, 110, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Lundstrom, K. RNA Viruses as Tools in Gene Therapy and Vaccine Development. Genes 2019, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Yan, G. The Identification and Development of a Novel Oncolytic Virus: Alphavirus M1. Hum. Gene Ther. 2021, 32, 138–149. [Google Scholar] [CrossRef]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-Tumor Interactome Screen Reveals ER Stress Response Can Reprogram Resistant Cancers for Oncolytic Virus-Triggered Caspase-2 Cell Death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Palese, P. Oncolytic Newcastle disease virus for cancer therapy: Old challenges and new directions. Future Microbiol. 2012, 7, 347–367. [Google Scholar] [CrossRef] [Green Version]
- Roshal, M.; Wood, B.L.; Fromm, J.R. Flow cytometric detection of the classical hodgkin lymphoma: Clinical and research applications. Adv. Hematol. 2011, 2011, 387034. [Google Scholar] [CrossRef]
- Nozawa, Y.; Wakasa, H.; Abe, M. Costimulatory molecules (CD80 and CD86) on Reed-Sternberg cells are associated with the proliferation of background T cells in Hodgkin’s disease. Pathol. Int. 1998, 48, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Zimpfer, A.; Pehrs, A.C.; Lugli, A.; Went, P.; Maurer, R.; Pileri, S.; Dirnhofer, S. Expression of B-cell markers in classical hodgkin lymphoma: A tissue microarray analysis of 330 cases. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2003, 16, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.; Thiesler, T.; Gerrits, B.; Behnke, S.; Sobotzki, N.; Omasits, U.; Bausch-Fluck, D.; Bock, T.; Aebersold, R.; Moch, H.; et al. Surfaceome of classical Hodgkin and non-Hodgkin lymphoma. Proteom. Clin. Appl. 2015, 9, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.S.; Weirather, J.L.; Lipschitz, M.; Lako, A.; Chen, P.H.; Griffin, G.K.; Armand, P.; Shipp, M.A.; Rodig, S.J. The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4-positive T cells that are PD-1-negative. Blood 2019, 134, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K. Contribution of Immunocytochemistry to the Diagnosis of Usual and Unusual Lymphoma Cases. J. Cytol. 2018, 35, 163–169. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Dhimolea, E. Brentuximab vedotin. mAbs 2012, 4, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.R.; Shipp, M.A. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.; Gandhi, M.K. Targeted Treatment of Follicular Lymphoma. J. Pers. Med. 2021, 11, 152. [Google Scholar] [CrossRef]
- Fessas, P.; Lee, H.; Ikemizu, S.; Janowitz, T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin. Oncol. 2017, 44, 136–140. [Google Scholar] [CrossRef]
- Voorhees, T.J.; Beaven, A.W. Therapeutic Updates for Relapsed and Refractory Classical Hodgkin Lymphoma. Cancers 2020, 12, 2887. [Google Scholar] [CrossRef]
- Vockerodt, M.; Cader, F.Z.; Shannon-Lowe, C.; Murray, P. Epstein-Barr virus and the origin of Hodgkin lymphoma. Chin. J. Cancer 2014, 33, 591–597. [Google Scholar] [CrossRef]
- Murray, P.G.; Young, L.S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Mackrides, N.; Campuzano-Zuluaga, G.; Maque-Acosta, Y.; Moul, A.; Hijazi, N.; Ikpatt, F.O.; Levy, R.; Verdun, R.E.; Kunkalla, K.; Natkunam, Y.; et al. Epstein-Barr virus-positive follicular lymphoma. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 2017, 30, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieser, A. Signal transduction by the Epstein-Barr virus oncogene latent membrane protein 1 (LMP1). Signal Transduct. 2007, 7, 20–33. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, P.G.; Al-Shamkhani, A. Soluble CD30 binds to CD153 with high affinity and blocks transmembrane signaling by CD30. Eur. J. Immunol. 2002, 32, 163–173. [Google Scholar] [CrossRef]
- Dong, L.; Hülsmeyer, M.; Dürkop, H.; Hansen, H.P.; Schneider-Mergener, J.; Ziegler, A.; Uchanska-Ziegler, B. Human CD30: Structural implications from epitope mapping and modeling studies. J. Mol. Recognit. 2003, 16, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Sci. 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Barh, D.; Tiwari, S.; Silva Andrade, B.; Giovanetti, M.; Almeida Costa, E.; Kumavath, R.; Ghosh, P.; Góes-Neto, A.; Carlos Junior Alcantara, L.; Azevedo, V. Potential chimeric peptides to block the SARS-CoV-2 spike receptor-binding domain. F1000Research 2020, 9, 576. [Google Scholar] [CrossRef]
- Oakley, B.R.; Paolillo, V.; Zheng, Y. γ-Tubulin complexes in microtubule nucleation and beyond. Mol. Biol. Cell 2015, 26, 2957–2962. [Google Scholar] [CrossRef]
- Wieczorek, M.; Urnavicius, L.; Ti, S.C.; Molloy, K.R.; Chait, B.T.; Kapoor, T.M. Asymmetric Molecular Architecture of the Human γ-Tubulin Ring Complex. Cell 2020, 180, 165–175.e116. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Feng, X.; Nie, L.; Tang, M.; Zhang, H.; Xiong, Y.; Swisher, S.K.; Srivastava, M.; Chen, J. Comprehensive analysis of the host-virus interactome of SARS-CoV-2. bioRxiv 2021, 12, 424961. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, H.T.; Shin, W.; Chae, J.; Choi, J.; Kim, S.H.; Lim, H.; Won Heo, T.; Park, K.Y.; Lee, Y.J.; et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 2016, 7, 13354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.; Zhang, H.; Chai, Y.; Song, H.; Tong, Z.; Wang, Q.; Qi, J.; Wong, G.; Zhu, X.; Liu, W.J.; et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 2017, 8, 14369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barh, D.; Tiwari, S.; Weener, M.E.; Azevedo, V.; Góes-Neto, A.; Gromiha, M.M.; Ghosh, P. Multi-omics-based identification of SARS-CoV-2 infection biology and candidate drugs against COVID-19. Comput. Biol. Med. 2020, 126, 104051. [Google Scholar] [CrossRef]
- Barh, D.; Tiwari, S.; Andrade, B.S.; Weener, M.E.; Góes-Neto, A.; Azevedo, V.; Ghosh, P.; Blum, K.; Ganguly, N.K. A novel multi-omics-based highly accurate prediction of symptoms, comorbid conditions, and possible long-term complications of COVID-19. Mol. Omics 2021, 17, 317–337. [Google Scholar] [CrossRef]
- Buckner, T.W.; Dunphy, C.; Fedoriw, Y.D.; van Deventer, H.W.; Foster, M.C.; Richards, K.L.; Park, S.I. Complete spontaneous remission of diffuse large B-cell lymphoma of the maxillary sinus after concurrent infections. Clin. Lymphoma Myeloma Leuk. 2012, 12, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Rudolphi-Solero, T.; Rashki, M.; Fernández-Fernández, J.; Rivas-Navas, D.; Ramos-Font, C.; Rodríguez-Fernández, A. SARS-COV-2 virus triggers immune antitumor response in a lymphoma patient. Rev. Esp. De Med. Nucl. E Imagen Mol. 2021. [Google Scholar] [CrossRef]
- O’Kelly, B.; McGettrick, P.; Angelov, D.; Fay, M.; McGinty, T.; Cotter, A.G.; Sheehan, G.; Lambert, J.S. Outcome of a patient with refractory Hodgkin lymphoma on pembrolizumab, infected with SARS-CoV-2. Br. J. Haematol. 2020, 190, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Han, J.; Liu, F.; Chen, X.; Qian, S.; Wang, Y.; Jia, Z.; Duan, X.; Zhang, X.; Zhu, J. Systematic Analysis of Coronavirus Disease 2019 (COVID-19) Receptor ACE2 in Malignant Tumors: Pan-Cancer Analysis. Front. Mol. Biosci. 2020, 7, 569414. [Google Scholar] [CrossRef] [PubMed]
- Regalado-Artamendi, I.A.; Jiménez-Ubieto, J.Á.; Hernández-Rivas, B.; Navarro, L.; Núñez, C.; Alaez, R.; Córdoba, F.J.; Peñalver, J.; Cannata, P.; Estival, K.; et al. Risk Factors and Mortality of COVID-19 in Patients with Lymphoma: A Multicenter Study. HemaSphere 2021, 5, e538. [Google Scholar] [CrossRef] [PubMed]
- Kanna, A.; Agrawal, S.; Jayant, K.; Kumar Pala, V.; Altujjar, M.; Hadid, T.; Khurram, M. B Cell Lymphoma, Unclassifiable, Transformed from Follicular Lymphoma: A Rare Presentation with Review of the Literature. Case Rep. Hematol. 2015, 2015, 651764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.Y.; Shahsafaei, A.; Dorfman, D.M. CD148 and CD27 are expressed in B cell lymphomas derived from both memory and naïve B cells. Leuk. Lymphoma 2002, 43, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Bosler, D.S.; Douglas-Nikitin, V.K.; Harris, V.N.; Smith, M.D. Detection of T-regulatory cells has a potential role in the diagnosis of classical Hodgkin lymphoma. Cytom. Part B Clin. Cytom. 2008, 74, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.S. Infrequent expression of CD15 by classical Hodgkin’s lymphomas in Taiwan. J. Clin. Pathol. 2017, 70, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, N.; Yagi, H.; Horibe, T.; Takigawa, M.; Sugiura, M. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma with a CD15(+)CD30(-) phenotype. Eur. J. Dermatol. 2007, 17, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Sano, D.; Hadid, T.H.; Singh, B.; Khan, U.; Kafri, Z.; Al-Katib, A.M. CD15 expression to predict outcome in patients with Hodgkin lymphoma. J. Clin. Oncol. 2016, 34, e19010. [Google Scholar] [CrossRef]
- Jones, R.J.; Gocke, C.D.; Kasamon, Y.L.; Miller, C.B.; Perkins, B.; Barber, J.P.; Vala, M.S.; Gerber, J.M.; Gellert, L.L.; Siedner, M.; et al. Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood 2009, 113, 5920–5926. [Google Scholar] [CrossRef] [Green Version]
- Ohana, O.M.; Ozer, J.; Prinsloo, I.; Benharroch, D.; Gopas, J. Hodgkin lymphoma cell lines bind to platelets. Incubation with platelets induces CD15 and P-selectin dependent adhesion of the cell lines to Human Umbilical Vein Endothelial cells (HUVEC). Cancer Biol. Ther. 2015, 16, 1651–1659. [Google Scholar] [CrossRef]
- Erlichman, B.; Zack Howard, O.M. CD27 signals through PKC in human B cell lymphomas. Cytokine 1999, 11, 476–484. [Google Scholar] [CrossRef]
- Vitale, L.A.; He, L.Z.; Thomas, L.J.; Widger, J.; Weidlick, J.; Crocker, A.; O’Neill, T.; Storey, J.; Glennie, M.J.; Grote, D.M.; et al. Development of a Human Monoclonal Antibody for Potential Therapy of CD27-Expressing Lymphoma and Leukemia. Clin. Cancer Res. 2012, 18, 3812–3821. [Google Scholar] [CrossRef] [Green Version]
- Gualco, G.; Natkunam, Y.; Bacchi, C.E. The spectrum of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma: A description of 10 cases. Mod. Pathol. 2012, 25, 661–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, P.; Díaz-Sámano, F.; Quiñonez, E. Diffuse Large B-Cell Lymphoma and Classical Hodgkin’S Lymphoma Converge in an Unusual Presentation as a Gastric Composite Lymphoma: Case Report. Arch. Hematol. Case Rep. Rev. 2016, 1, 001–002. [Google Scholar] [CrossRef] [Green Version]
- Perron, M.; Saragovi, H.U. Inhibition of CD45 Phosphatase Activity Induces Cell Cycle Arrest and Apoptosis of CD45+ Lymphoid Tumors Ex Vivo and In Vivo. Mol. Pharmacol. 2018, 93, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.C.; Lahtz, T.; Nagao, J.Y.; Song, W.C.; Chan, H.; Lee, C.; Yue, T.; Look, R.; Mülfarth, W.; Li, K.; et al. CTLA4 Promotes Tyk2-STAT3–Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanina, N.; Kline, J.; Bishop, M.R. Emerging role of checkpoint blockade therapy in lymphoma. Ther. Adv. Hematol. 2017, 8, 81–90. [Google Scholar] [CrossRef]
- Hollander, P.; Amini, R.-M.; Ginman, B.; Molin, D.; Enblad, G.; Glimelius, I. Expression of PD-1 and PD-L1 increase in consecutive biopsies in patients with classical Hodgkin lymphoma. PLoS ONE 2018, 13, e0204870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravelle, P.B.; Burroni, S.; Péricart, C.; Rossi, C.; Bezombes, M.; Tosolini, D.; Damotte, P.; Brousset, J.-J.; Fournié, L.C. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: A summary of immunohistochemical studies. Oncotarget 2017, 8, 44960–44975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; Popa McKiver, M.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef]
- Gioti, K.; Kottaridi, C.; Voyiatzaki, C.; Chaniotis, D.; Rampias, T.; Beloukas, A. Animal Coronaviruses Induced Apoptosis. Life 2021, 11, 185. [Google Scholar] [CrossRef]
- Chu, H.; Shuai, H.; Hou, Y.; Zhang, X.; Wen, L.; Huang, X.; Hu, B.; Yang, D.; Wang, Y.; Yoon, C.; et al. Targeting highly pathogenic coronavirus-induced apoptosis reduces viral pathogenesis and disease severity. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.Y.; Yeung, Y.S.; Hon, C.C.; Zeng, F.; Law, K.M.; Leung, F.C. SARS coronavirus and apoptosis. Hong Kong Med. J. 2008, 14, 8–13. [Google Scholar] [PubMed]
- Zhao, G.; Shi, S.Q.; Yang, Y.; Peng, J.P. M and N proteins of SARS coronavirus induce apoptosis in HPF cells. Cell Biol. Toxicol. 2006, 22, 313–322. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, F.J.; Moreno-Artero, E.; Jahnke, S. SARS-CoV-2 and EBV coinfection. Med. Clin. 2020, 155, 319–320. [Google Scholar] [CrossRef]
- Paolucci, S.; Cassaniti, I.; Novazzi, F.; Fiorina, L.; Piralla, A.; Comolli, G.; Bruno, R.; Maserati, R.; Gulminetti, R.; Novati, S.; et al. EBV DNA increase in COVID-19 patients with impaired lymphocyte subpopulation count. Int. J. Infect. Dis. 2021, 104, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Song, J.; Liu, H.; Zheng, H.; Chen, C. Positive Epstein-Barr virus detection in coronavirus disease 2019 (COVID-19) patients. Sci. Rep. 2021, 11, 10902. [Google Scholar] [CrossRef]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef] [PubMed]
- Roncati, L.; Lusenti, B.; Nasillo, V.; Manenti, A. Fatal SARS-CoV-2 coinfection in course of EBV-associated lymphoproliferative disease. Ann. Hematol. 2020, 99, 1945–1946. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | % of Residues in | |||
|---|---|---|---|---|
| Most Favored Regions | Additional Allowed Regions | Generously Allowed Regions | Disallowed Regions | |
| Human CD30 | 87.0 | 11.0 | 1.4 | 0.6 |
| Human CD15 | 92.0 | 7.5 | 0.0 | 0.5 |
| EBV LMP-1 | 96.0 | 4.0 | 0.0 | 0.0 |
| SASR-CoV-2 M protein | 92.5 | 7.0 | 0.5 | 0.0 |
| Protein Complexes | Docked Complex Number | Interacting Residues |
|---|---|---|
| CD15- Spike RBD | 3 | Trp-500-Tyr505, Arg428-Thr500, Glu502-Gln498, Glu-502-Tyr449, Trp504-Gly446, Asp-210-Tyr453, Pro207-Ser494, Arg206-Tyr449, Arg206-Ser494, Arg206-Asn450, Leu178-Tyr489, Leu178-Asn487 |
| 4 | Gly340-Tyr449, Ser344-Gln498, Ser344-Gly496, Ser344-Asn501, Arg449-Asn487, Leu333-Tyr489, Glu448-Tyr473, Ser337-Ser494, Ser337-Leu492, Arg336-Gln493 | |
| 6 | Arg214-Gly496, Asp210-Tyr449, Arg212-Glu484, Ser498-Gly446, Leu178-Lys417, Gln174-Asp405, Tyr169-Tyr505, Cys171-Gly502 | |
| CD27- Spike RBD | 8 | Ile46-Gln498, Ser50-Gln498, Thr57-Gln493, Leu29-Tyr453, His60-Tyr495, Cys61-Tyr449, Gln15-Tyr421, Trp13-Ala475, Trp13-Tyr489 |
| 9 | Arg87-Tyr495, Trp90-Gln498, Asn88-Gln498, Asn88-Thr500, Gly70-Gln493, Leu72-Tyr453, Leu72-Arg403, Ser63-Asn487, Ser63-Tyr489, Gly48-Lys417 | |
| CD45- Spike RBD | 3 | Lys210-Glu484, His191-Cys488, Ile235-Asn487, Lys157-Tyr473, Lys157-Arg457, Pro186-Gln493, Gln187-Arg403, His134-Asn460, Asn184-Gln498 |
| 8 | Ile235-Asn487, His191-Cys488, Pro186-Gln493, Gln187-Tyr453, Asn184-Gln498, Gln167-Asn501 | |
| CD152- Spike RBD | 2 | Asp118-Tyr449, Asp118-Gly496, Pro119-Gln498, Gly111-Asn487, Ala86-Gln493, Met87-Ser494, Ala42-Tyr421, Gln45-Asp420, Gln45-Tyr421, Asp43-Arg457 |
| 3 | Met87-Gln498, Asp88-Asn501, Asp88-Thr500, Arg40-Gly446, Tyr104-Tyr489, Ile67-Tyr505, Glu48-Tyr453, Thr47-Ser494, Val46-Ser494 |
| Protein Complexes | Docked Complex Number | Interacting Residues |
|---|---|---|
| GCP2-CoV-2 M Protein (Lateral Position) | 7 | Arg290-Cys33, Arg290-Thr30, Trp353-Thr30, Trp353-Phe26, Gln350-Lys50, Ser283-Asn43 |
| 3 | Ser243-Ser173, Ser243-Arg174, Thr235-Arg198, Arg290-Leu56, Ser393-Asn5, Ser393-Thr7 | |
| GCP2- Orf3a (Lateral Position) | 4 | Arg361-Asp210, Ser393-Ile236, Glu242-Lys61, Asp397-Ser209, Trp353-Asp142, Arg290-Tyr189 |
| GCP3-Orf3a (Lateral Position) | 7 | Arg333-Asp142, Arg252-Ser165, Ser323-Lys61, Gln322-Lys61, His343-Asp210 |
| 3 | Tyr256-Val163, Arg252-Val163, Arg337-Ile118, Arg333-Asn119, Gln416-Tyr107, Arg315-Lys61, Gln322-Ser58 | |
| GCP2-M Protein (At GCP2 residues that interact with TUBG1) | 5 | Asp554-Lys205, Tyr723-Asn117, Arg711-Glu137, Asp561-Thr208, Ser885-Arg101 |
| 1 | Ser885-Arg105, Ser885-Arg101, Cys684-Cys86, Glu731-Gln36 | |
| GCP3-M Protein (At GCP3 residues that interact with TUBG1) | 1 | Phe612-Gln36, Gln570-Gln36, Ser709-Arg131, Ser709-His154, Lys689-Glu137, Thr678-Arg107, Glu728-Arg42, Gly569-Arg42, Tyr720-Asn41, Lys671-Tyr39 |
| 5 | Phe612-Gln36, Arg681-Glu137, Glu728-Arg42, Ser709-His154, His716-Arg107, Asn883-His155, Gly571-Arg105, Asp572-Arg105 | |
| GCP2-Orf3a (At GCP2 residues that interact with TUBG1) | 3 | Cys684-Val163, Gln719-Ser166, Tyr723-Ser166, Asn890-Tyr215 |
| 9 | Gln722-Lys192, Gln719-Trp193, Cys684-His182, Asn716-Cys153, Gln712-Asn152, Asp561-His227, Arg681-Gly174 | |
| GCP3-Orf3a (At GCP3 residues that interact with TUBG1 | 1 | His716-Asp210, Tyr720-Asp210, Asn883-Asn144, Gln717-Tyr145, Ser709-Ser162, Arg681-Ser166, Arg681-Thr164, Thr678-Ser165 |
| 3 | His702-Leu83, Glu728-Lys192, Asn609-Thr151, Gln570-Asn152, Gln717-Trp69 | |
| TUBG1-M Protein (At TUBG1 residues that interact with GCP2) | 1 | Pro350-Arg131, Trp351-Arg131, Asp442-His125, Arg341-Leu134, Asn251-Gln36, His334-Arg101, Tyr248-Tyr47, Asp252-Arg42 |
| 7 | Leu276-Arg44, Tyr273-Arg42, Lys363-Phe37, Glu38-Arg105, Ser32-Arg101, Lys23-Arg105, Ser80-107, Thr234-Tyr39, Thr234-Asn41 | |
| 8 | Glu426-Arg131, Pro162-Arg42, Asp200-Arg42, Gln197-Trp110, Pro-264-Tyr39, Thr196-Asn41, Arg265-Asn41, Asp433-Arg107, Asp419-Thr127, Asp422-His125 | |
| TUBG1-M Protein (At TUBG1 residues that interact with GCP3) | 1 | Pro350-Arg131, Trp351- Arg131, Asp442-His125, Arg341-Leu134, Asn251-Gln36, His334-Arg101, Tyr248-Tyr47, Asp252-Arg42 |
| 8 | Glu426-Arg131, Pro162-Arg42, Asp200-Arg42, Gln197-Trp110, Pro-264-Tyr39, Thr196-Asn41, Arg265-Asn41, Asp433-Arg107, Asp419-Thr127, Asp422-His125 | |
| 9 | Gln357-Thr127, Thr331-Arg131, His334-Leu156, Ser355-His125, Arg341-Asp160, Glu327-Asn41 | |
| TUBG1-orf3a (At TUBG1 residues that interact with GCP2) | 3 | Val130-Lys136, Arg212-Tyr154, Arg390-Arg134, Arg390-Asp134, Asp216-Arg68, Arg343-Leu83, Gln394-Asn152 |
| 1 | Arg341-Phe207, His334-Tyr211, Gln357-Ile124, Tyr248-Cys130 | |
| TUBG1-orf3a (At TUBG1 residues that interact with GCP3) | 1 | Arg341-Phe207, His334-Tyr211, Gln357-Ile124, Tyr248-130 |
| 3 | Val305-Lys136, Arg212-Tyr154, Arg390-Asp155, Arg390-Arg134, Asp216-Arg68, Arg343-Leu83, Gln394-Asn152 |
| Protein Complexes | Docked Complex Number | Interacting Residues |
|---|---|---|
| LMP-1 and 3CLpro/Mpro | 1 | His352-Asn274, Tyr384-Asn274, Tyr385-Gly275, Leu382-Asn277, Ser383-Asn277, Ser367-Ala285 |
| 9 | Ser383-Asn277, Tyr385-Arg279, Tyr384-Glu270, Ser313-Glu270, Ser313-Lys269, Thr324-Gln273, Glu328-Lys236 | |
| 10 | Gln381-Arg298, Gln381-Gly302, Gln381-Ser301, Gly345-Thr304, Asp341-Gln306, Asp341-Phe305, Ser347-Ser1, Ser383-Ser1, Val228-Arg4, Ala231-Phe3 | |
| LMP-1 and NSP7 | 6 | Ser383-Leu60, Tyr385-Leu59, Tyr385-Met62, Ser367-Met62, Ser367-Leu20, Gly368-Leu20, Gly371-Arg21, Ser369-Ser24 |
| 7 | Tyr384-Gly64, Ser313-Lys70, Tyr385-Leu59, Tyr385-Met62, Gln381-Ser61, Ser367-Ser61, Ser367-Val58, Gly371-Ser26 | |
| 8 | Ser313-Lys70, Ser367-Leu20, Tyr385-Met62, Tyr385-Leu59, Tyr385-Val66, Ser383-Leu60, Gln381-Ser61 | |
| LMP-1 and NSP10 | 2 | Ser313-Asp65, Ser383-His63, Gln381-His63, Tyr384-His63, Tyr385-His63, Ser367-Arg61, Gly365-Cys62, Glu325-Asp5 |
| 8 | Tyr385-Ser112, Tyr385-Arg117, Ser383-Arg117, Tyr384-Cys113, Gln381-Glu118, Ser347-Leu121, His346-Gln122, Thr324-Val102 | |
| LMP-1 and Spike RBD | 6 | His352-Tyr489, Gln381-Gln493, Tyr384-Tyr489, Tyr384-Asn487, Tyr385-Lys417 |
| 7 | Tyr384-Asn487, His352-Asn487, His352-Tyr489, Asp386-Tyr421, Ser367-Leu492, Ser367-Phe490, Ser367-Gln493 | |
| 8 | Pro376-Asn501, Pro376-Gln498, Gly344-Tyr453, Asp341-Lys417, Tyr385-Phe486, Ser229-Asn487, Gln381-Glu484 | |
| PD-1 and Spike RBD | 2 | Thr53-Tyr505, His107-Tyr505, Arg104-Tyr505, Ser55-Tyr453, Thr36-Tyr453, Glu136-Tyr489, Lys135-Cys488, Trp32-Glu484, Asn58-Tyr449 |
| 7 | Ser127-Tyr489, Ala129-Tyr489, Leu100-Tyr449, Pro101-Gln498, Phe63-Gln493, Tyr68-Glu484 | |
| 8 | Asp77-Gln493, Lys78-Phe490, Arg94-Tyr505, Ile126-Phe486, Gln75-Tyr449, Ser71-Tyr449, Ser71-Gly446, Ser73-Gly446 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barh, D.; Tiwari, S.; Gabriel Rodrigues Gomes, L.; Weener, M.E.; Alzahrani, K.J.; Alsharif, K.F.; Aljabali, A.A.A.; Tambuwala, M.M.; Lundstrom, K.; Hassan, S.S.; et al. Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas. Viruses 2021, 13, 1927. https://doi.org/10.3390/v13101927
Barh D, Tiwari S, Gabriel Rodrigues Gomes L, Weener ME, Alzahrani KJ, Alsharif KF, Aljabali AAA, Tambuwala MM, Lundstrom K, Hassan SS, et al. Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas. Viruses. 2021; 13(10):1927. https://doi.org/10.3390/v13101927
Chicago/Turabian StyleBarh, Debmalya, Sandeep Tiwari, Lucas Gabriel Rodrigues Gomes, Marianna E. Weener, Khalid J. Alzahrani, Khalaf F. Alsharif, Alaa A. A. Aljabali, Murtaza M. Tambuwala, Kenneth Lundstrom, Sk. Sarif Hassan, and et al. 2021. "Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas" Viruses 13, no. 10: 1927. https://doi.org/10.3390/v13101927
APA StyleBarh, D., Tiwari, S., Gabriel Rodrigues Gomes, L., Weener, M. E., Alzahrani, K. J., Alsharif, K. F., Aljabali, A. A. A., Tambuwala, M. M., Lundstrom, K., Hassan, S. S., Serrano-Aroca, Á., Takayama, K., Ghosh, P., Redwan, E. M., Silva Andrade, B., Soares, S. d. C., Azevedo, V., & Uversky, V. N. (2021). Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas. Viruses, 13(10), 1927. https://doi.org/10.3390/v13101927