
Primary HSV-2 Infection in Early Pregnancy Results in Transplacental Viral Transmission and Dose-Dependent Adverse Pregnancy Outcomes in a Novel Mouse Model


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Primary HSV-2 Inoculation
2.3. Vaginal Washes and Pathology Scoring
2.4. Viral Titers
2.5. Viral DNA Extraction
2.6. Quantification of HSV-2 Viral DNA
2.7. Histology and Morphometry
2.7.1. Hematoxylin and Eosin (H&E) Staining
2.7.2. HSV-2 Immunohistochemistry (IHC)
2.8. Tissue Homogenization and Multiplex Cytokine/Chemokine Analysis
2.9. Statistical Analysis
3. Results
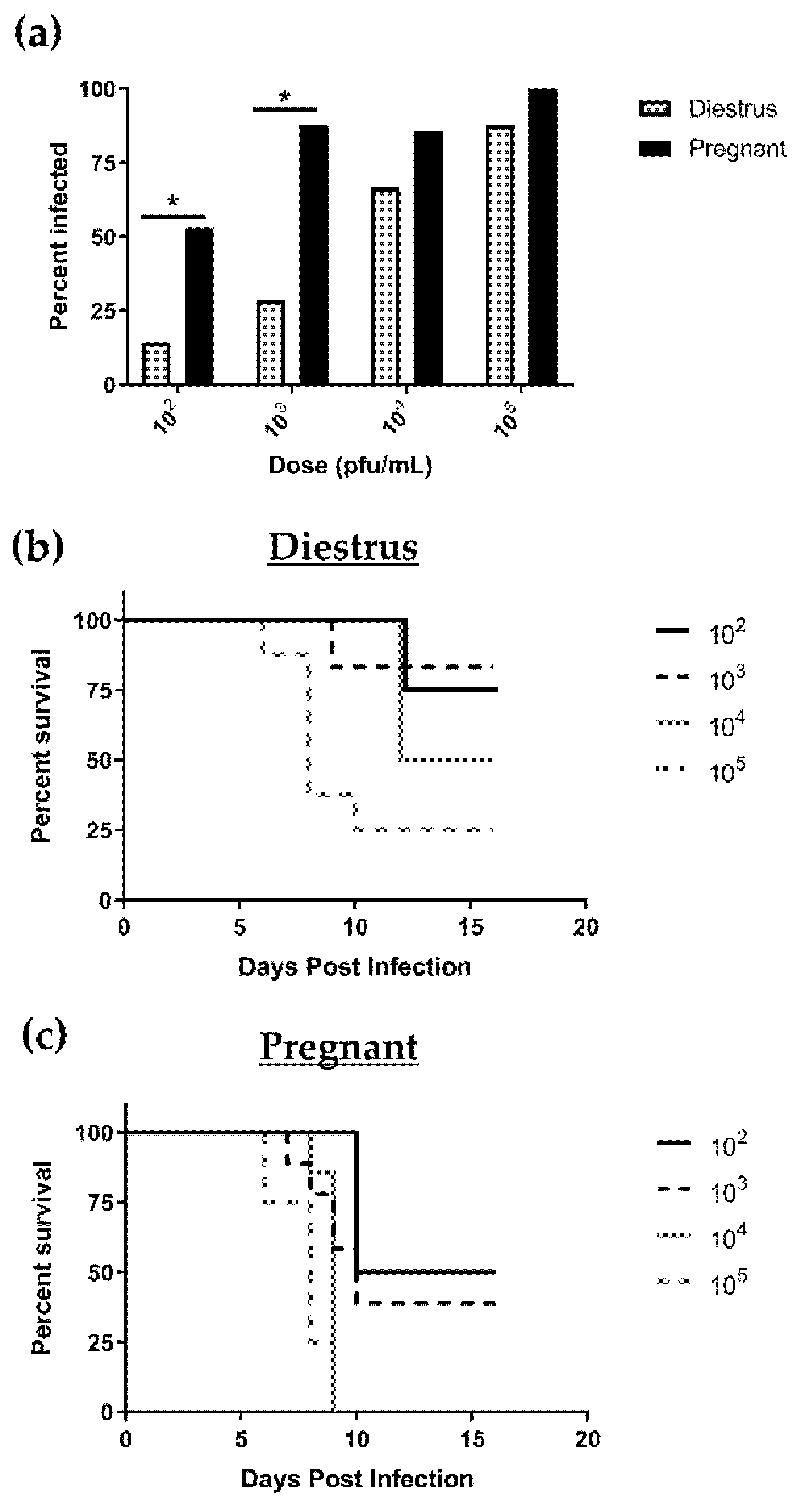
3.1. Pregnant Mice Are 100 Times More Susceptible to Primary HSV-2 Infection in Early Pregnancy than Nonpregnant, Diestrus-Staged Controls
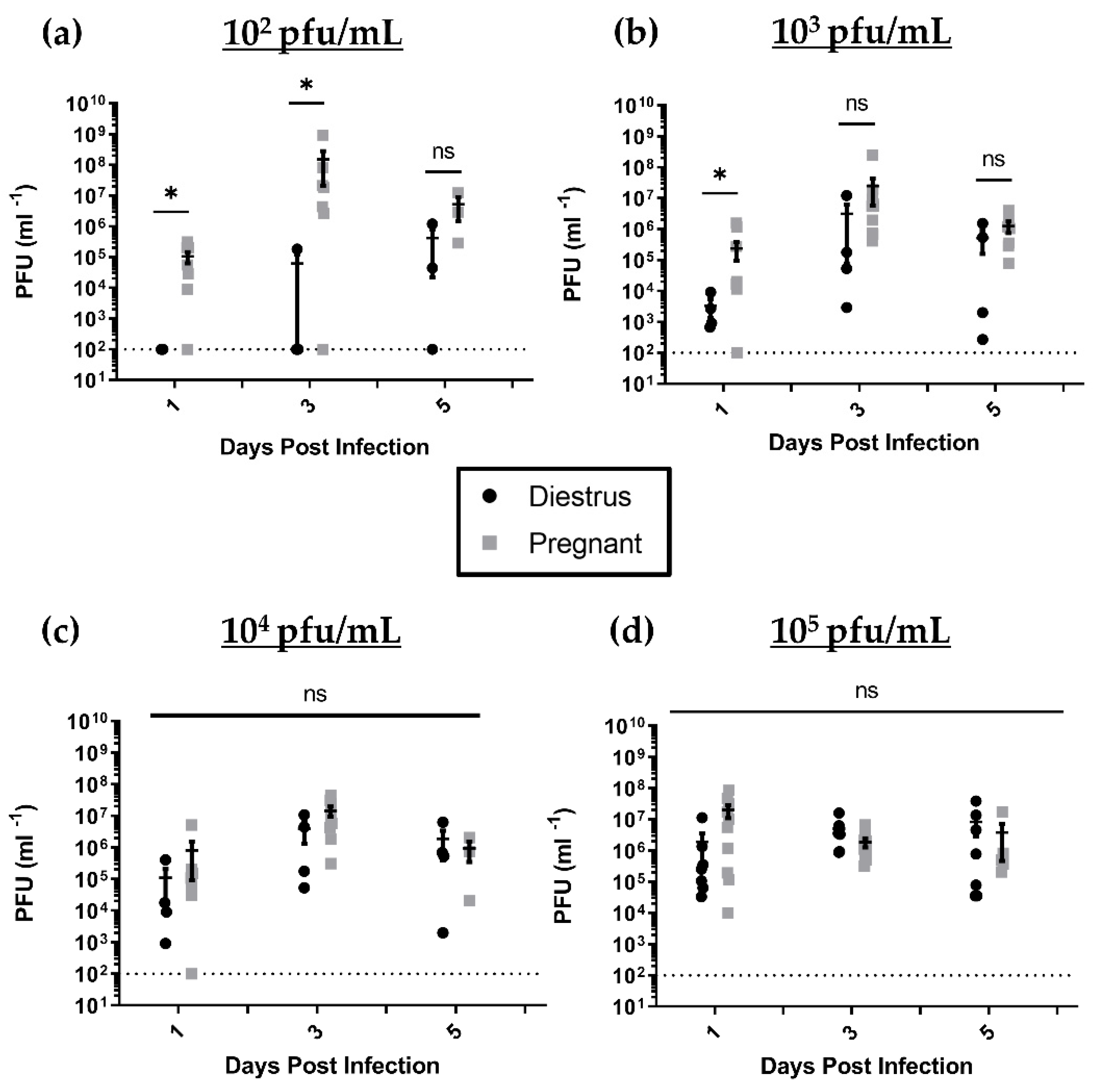
3.2. HSV-2-Infected Pregnant Mice Display Higher Genital Pathology and Enhanced Viral Shedding Compared to Nonpregnant, Diestrus-staged Controls
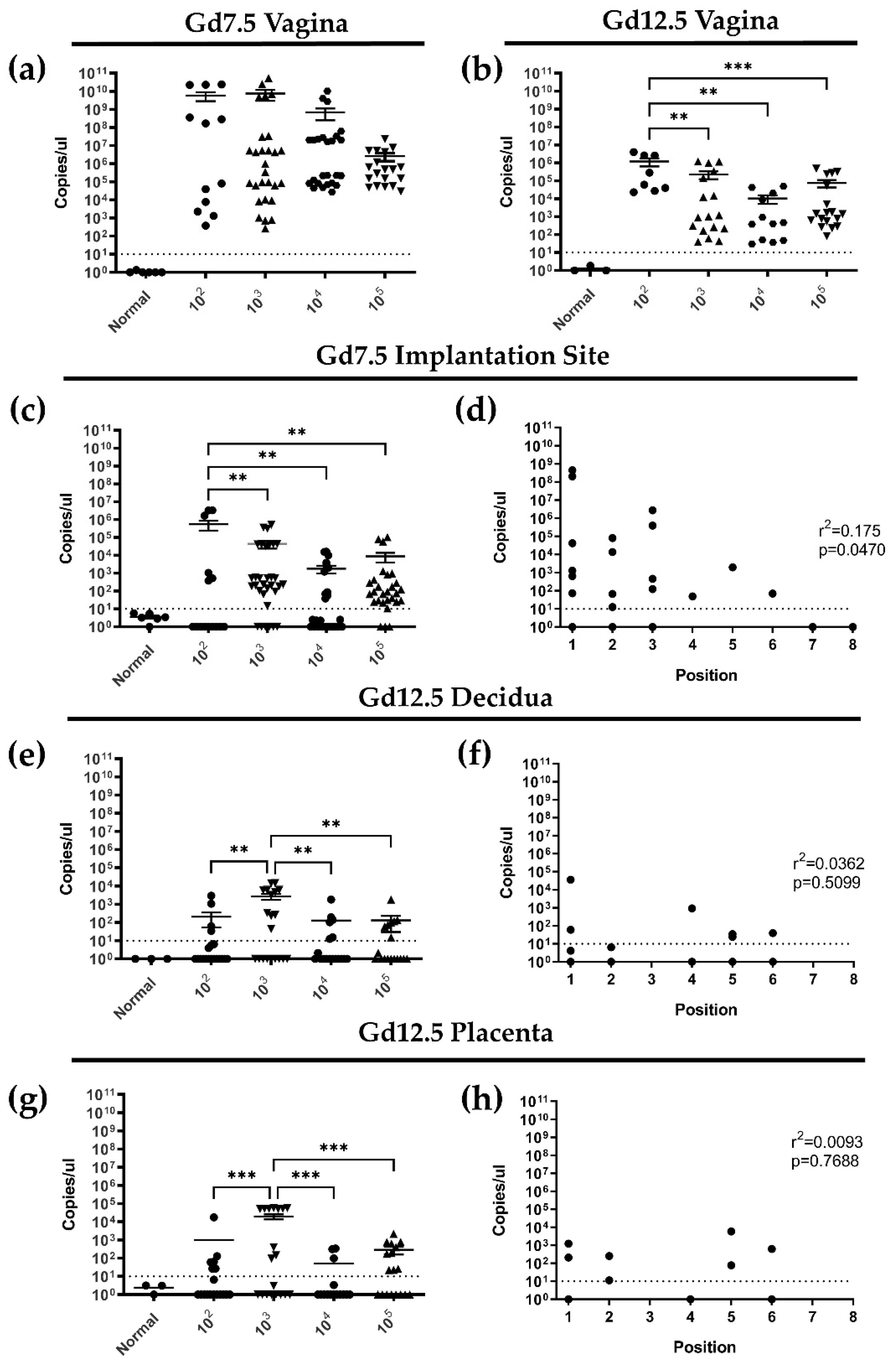
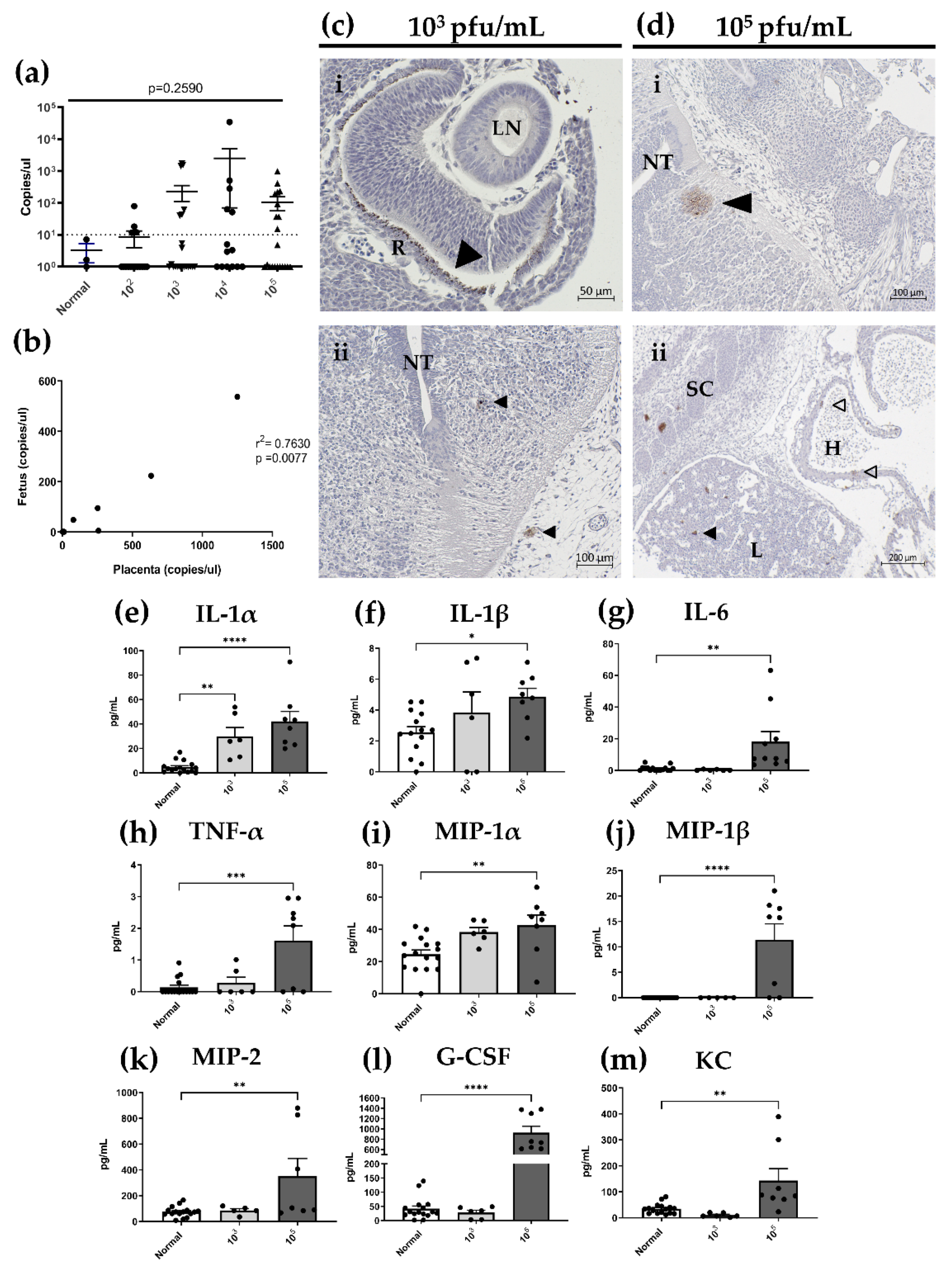
3.3. Following Primary Intravaginal Infection in Early Pregnancy, HSV-2 Ascends from the Vaginal Tract in a Directional Manner and HSV-2 DNA Is Detectable in Implantation Sites at gd7.5 and 12.5
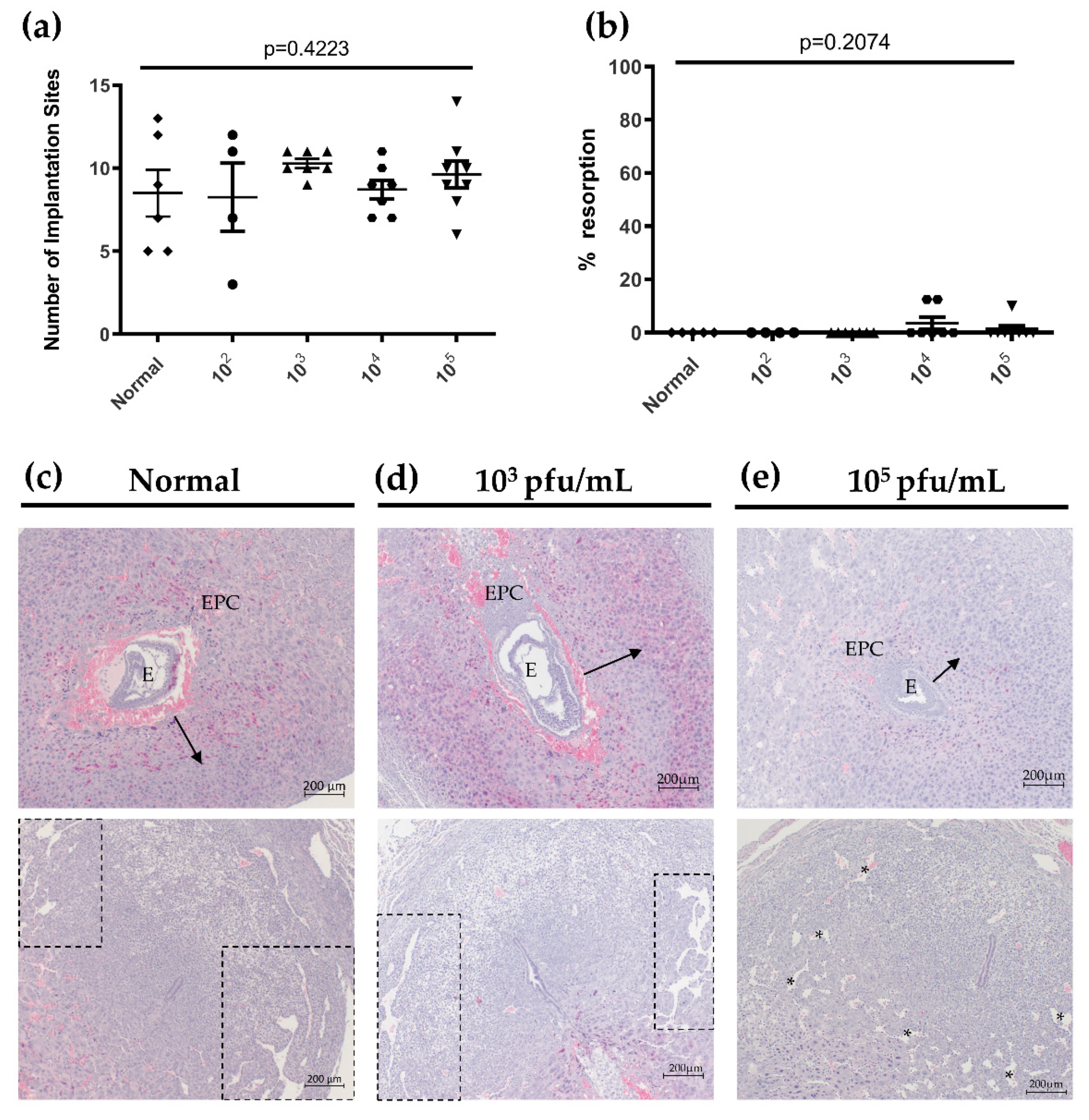
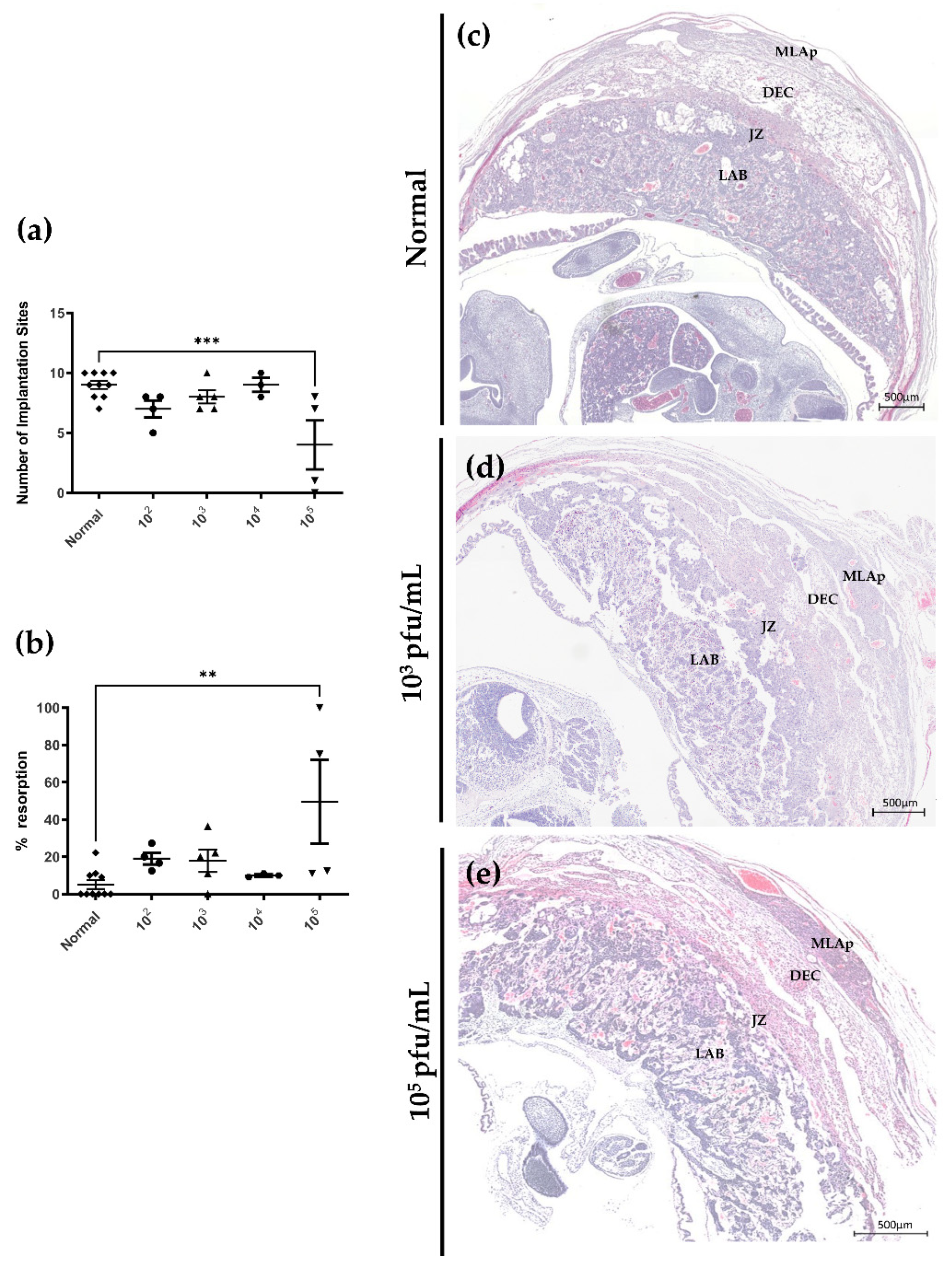
3.4. Primary HSV-2 Infection in Early Pregnancy Affects Fetal Outcomes at gd12.5, but Not at gd 7.5
3.5. HSV-2 Infection of Maternal and Fetal Placental Tissues Correlates with Histopathology Consistent with Loss of Integrity and Decreased Fetal Growth at gd12.5
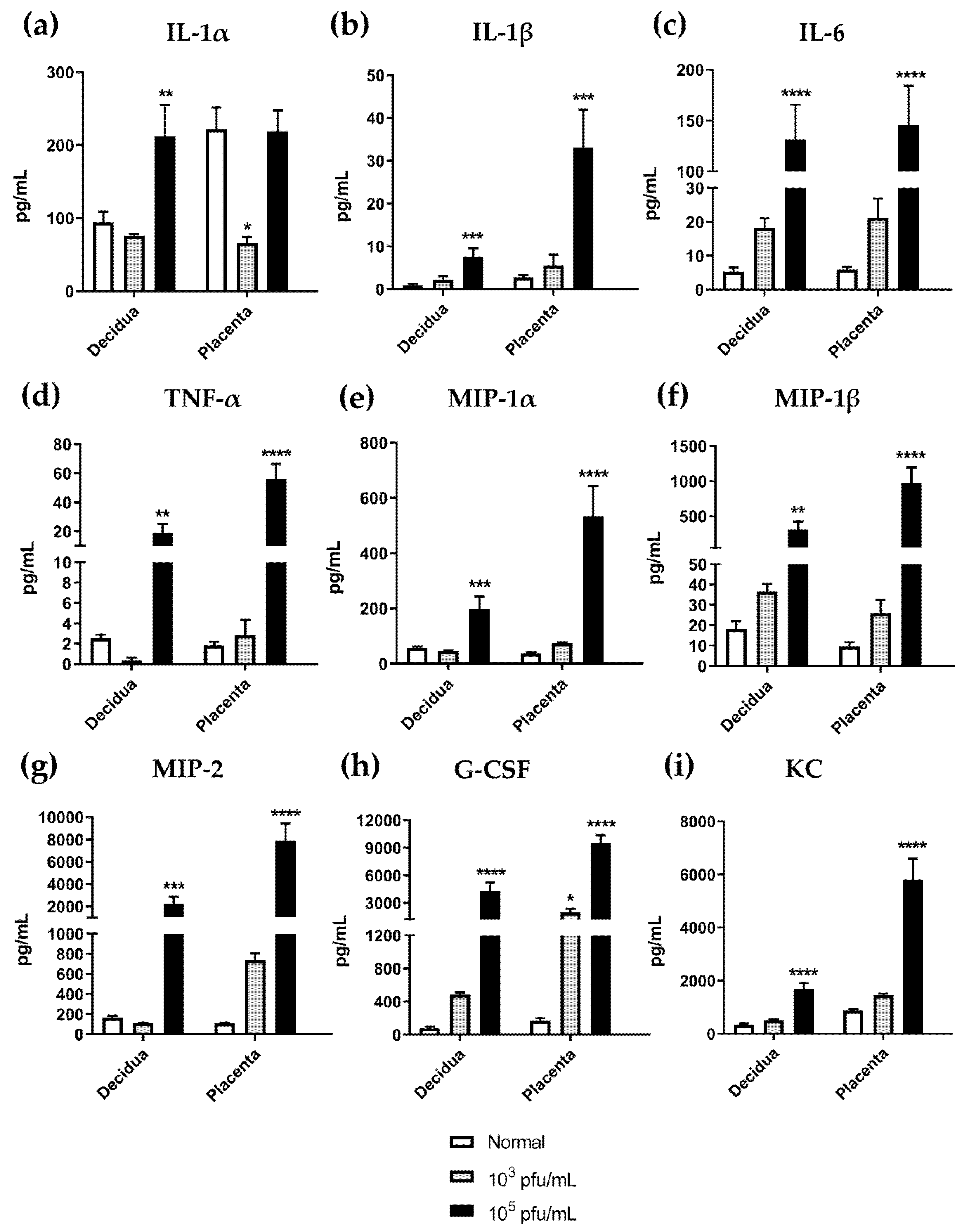
3.6. High Dose HSV-2 Infection Is Associated with Increased Levels of Pro-Inflammatory Cytokines in Decidual and Placental Tissues on gd12.5
3.7. In Utero HSV-2 Transmission into Fetuses Is Observed at gd12.5 and Is Accompanied by Inflammatory Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Kaushic, C.; Ashkar, A.A.; Reid, L.A.; Rosenthal, K.L. Progesterone increases susceptibility and decreases immune responses to genital herpes infection. J. Virol. 2003, 77, 4558–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillgrass, A.E.; Fernandez, S.A.; Rosenthal, K.L.; Kaushic, C. Estradiol regulates susceptibility following primary exposure to genital herpes simplex virus type 2, while progesterone induces inflammation. J. Virol. 2005, 79, 3107–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teepe, A.G.; Allen, L.B.; Wordinger, R.J.; Harris, E.F. Effect of the estrous cycle on susceptibility of female mice to intravaginal inoculation of herpes simplex virus type 2 (HSV-2). Antiviral Res. 1990, 14, 227–235. [Google Scholar] [CrossRef]
- Lee, Y.; Dizzell, S.E.; Leung, V.; Nazli, A.; Zahoor, M.A.; Fichorova, R.N.; Kaushic, C. Effects of Female Sex Hormones on Susceptibility to HSV-2 in Vaginal Cells Grown in Air-Liquid Interface. Viruses 2016, 8, 241. [Google Scholar] [CrossRef]
- Hammad, W.A.B.; Konje, J.C. Herpes simplex virus infection in pregnancy—An update. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 259, 38–45. [Google Scholar] [CrossRef]
- Silasi, M.; Cardenas, I.; Kwon, J.-Y.; Racicot, K.; Aldo, P.; Mor, G. Viral infections during pregnancy. Am. J. Reprod. Immunol. 2015, 73, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.J.; Embleton, N.D.; Clark, J.E.; Bythell, M.; Ward Platt, M.P.; Berrington, J.E. Viral infections: Contributions to late fetal death, stillbirth, and infant death. J. Pediatr. 2013, 163, 424–428. [Google Scholar] [CrossRef]
- Auriti, C.; De Rose, D.U.; Santisi, A.; Martini, L.; Piersigilli, F.; Bersani, I.; Ronchetti, M.P.; Caforio, L. Pregnancy and Viral infections: Mechanisms of fetal damage, diagnosis and prevention of neonatal adverse outcomes from cytomegalovirus to SARS-CoV-2 and Zika virus. Biochim. Biophys. Acta. Mol. Basis. Dis. 2021, 166198. [Google Scholar] [CrossRef]
- James, S.H.; Sheffield, J.S.; Kimberlin, D.W. Mother-to-Child Transmission of Herpes Simplex Virus. J. Pediatric. Infect. Dis. Soc. 2014, 3 (Suppl. 1), S19–S23. [Google Scholar] [CrossRef] [Green Version]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Newman, L.M.; Gottlieb, S.L. First estimates of the global and regional incidence of neonatal herpes infection. Lancet. Glob. Health 2017, 5, e300–e309. [Google Scholar] [CrossRef] [Green Version]
- Stephenson-Famy, A.; Gardella, C. Herpes simplex virus infection during pregnancy. Obstet. Gynecol. Clin. N. Am. 2014, 41, 601–614. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. Management of Genital Herpes in Pregnancy: ACOG Practice Bulletinacog Practice Bulletin, Number 220. Obstet. Gynecol. 2020, 135, e193–e202. [Google Scholar] [CrossRef] [PubMed]
- Money, D.M.; Steben, M. No. 208-Guidelines for the Management of Herpes Simplex Virus in Pregnancy. J. Obstet. Gynaecol. Can. 2017, 39, e199–e205. [Google Scholar] [CrossRef]
- Kim, I.D.; Chang, H.S.; Hwang, K.J. Herpes simplex virus 2 infection rate and necessity of screening during pregnancy: A clinical and seroepidemiologic study. Yonsei. Med. J. 2012, 53, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalu, E.I.; Ojide, C.K.; Chuku, A.; Chukwuonye, I.I.; Agwu, F.E.; Nwadike, V.U.; Korie, F.C.; Okafor, G.O.C. Obstetric outcomes of human herpes virus-2 infection among pregnant women in Benin, Nigeria. Niger. J. Clin. Pract. 2015, 18, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.L.; Huang, L.J.; Xiong, Y.Q.; Zhong, Y.Y.; Yang, J.J.; Fu, T.; Lei, X.F.; Chen, Q. The risk of herpes simplex virus and human cytomegalovirus infection during pregnancy upon adverse pregnancy outcomes: A meta-analysis. J. Clin. Virol. 2018, 104, 48–55. [Google Scholar] [CrossRef]
- Brown, Z.A.; Vontver, L.A.; Benedetti, J.; Critchlow, C.W.; Sells, C.J.; Berry, S.; Corey, L. Effects on infants of a first episode of genital herpes during pregnancy. N. Engl. J. Med. 1987, 317, 1246–1251. [Google Scholar] [CrossRef]
- Longo, S.; Borghesi, A.; Tzialla, C.; Stronati, M. IUGR and infections. Early Hum. Dev. 2014, 90 (Suppl. 1), S42–S44. [Google Scholar] [CrossRef]
- McGee, D.; Smith, A.; Poncil, S.; Patterson, A.; Bernstein, A.I.; Racicot, K. Cervical HSV-2 infection causes cervical remodeling and increases risk for ascending infection and preterm birth. PLoS ONE 2017, 12, e0188645. [Google Scholar] [CrossRef] [Green Version]
- Bhatta, A.K.; Keyal, U.; Liu, Y.; Gellen, E. Vertical transmission of herpes simplex virus: An update. J. Dtsch. Dermatol. Ges. 2018, 16, 685–692. [Google Scholar] [CrossRef]
- Samies, N.L.; James, S.H. Prevention and treatment of neonatal herpes simplex virus infection. Antiviral Res. 2020, 176, 104721. [Google Scholar] [CrossRef]
- Finger-Jardim, F.; Avila, E.C.; da Hora, V.P.; Gonçalves, C.V.; de Martinez, A.M.B.; Soares, M.A. Prevalence of herpes simplex virus types 1 and 2 at maternal and fetal sides of the placenta in asymptomatic pregnant women. Am. J. Reprod. Immunol. 2017, 78, e12689. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J.; Fowler, K.; Hutto, S.C.; James, S.H.; Kimberlin, D.W.; Poole, C.; Ross, S.A.; Whitley, R.J. Pathogenesis of Non-Zika Congenital Viral Infections. J. Infect. Dis. 2017, 216, S912–S918. [Google Scholar] [CrossRef] [Green Version]
- Bibbins-Domingo, K.; Grossman, D.C.; Curry, S.J.; Davidson, K.W.; Epling, J.W., Jr.; García, F.A.; Kemper, A.R.; Krist, A.H.; Kurth, A.E.; Landefeld, C.S.; et al. Serologic Screening for Genital Herpes Infection: US Preventive Services Task Force Recommendation Statement. JAMA 2016, 316, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.M.; Shekhawat, P.; Reynolds, A.S.; Thakur, K.T. Neurologic infections during pregnancy. Handb. Clin. Neurol. 2020, 172, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, N.A.; Zimberlin, M.N. Pathogenesis of herpes simplex virus type 2 experimental genital infection in pregnant mice. FEMS Immunol. Med. Microbiol. 2001, 30, 197–202. [Google Scholar] [CrossRef] [PubMed]
- LaTourette, P.C., 2nd; Awasthi, S.; Desmond, A.; Pardi, N.; Cohen, G.H.; Weissman, D.; Friedman, H.M. Protection against herpes simplex virus type 2 infection in a neonatal murine model using a trivalent nucleoside-modified mRNA in lipid nanoparticle vaccine. Vaccine 2020, 38, 7409–7413. [Google Scholar] [CrossRef]
- Wood, G.A.; Fata, J.E.; Watson, K.L.; Khokha, R. Circulating hormones and estrous stage predict cellular and stromal remodeling in murine uterus. Reproduction 2007, 133, 1035–1044. [Google Scholar] [CrossRef]
- Thaete, L.G.; Levin, S.I.; Dudley, A.T. Impact of anaesthetics and analgesics on fetal growth in the mouse. Lab. Anim. 2013, 47, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Bagri, P.; Anipindi, V.C.; Nguyen, P.V.; Vitali, D.; Stämpfli, M.R.; Kaushic, C. Novel Role for Interleukin-17 in Enhancing Type 1 Helper T Cell Immunity in the Female Genital Tract following Mucosal Herpes Simplex Virus 2 Vaccination. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Vitali, D.; Bagri, P.; Wessels, J.M.; Arora, M.; Ganugula, R.; Parikh, A.; Mandur, T.; Felker, A.; Garg, S.; Kumar, M.; et al. Curcumin Can Decrease Tissue Inflammation and the Severity of HSV-2 Infection in the Female Reproductive Mucosa. In.t J. Mol. Sci. 2020, 21, 337. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Y.; Zou, H.; Ong, D.C.; Ker, K.J.; Chio, M.T.; Teo, R.Y.; Koh, M.J. Development and clinical validation of a multiplex real-time PCR assay for herpes simplex and varicella zoster virus. Diagn. Mol. Pathol. 2013, 22, 245–248. [Google Scholar] [CrossRef]
- Felker, A.M.; Chen, Z.; Foster, W.G.; Croy, B.A. Receptors for non-MHC ligands contribute to uterine natural killer cell activation during pregnancy in mice. Placenta 2013, 34, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croy, B.A.; Chen, Z.; Hofmann, A.P.; Lord, E.M.; Sedlacek, A.L.; Gerber, S.A. Imaging of vascular development in early mouse decidua and its association with leukocytes and trophoblasts. Biol. Reprod. 2012, 87, 125. [Google Scholar] [CrossRef]
- Adamson, S.L.; Lu, Y.; Whiteley, K.J.; Holmyard, D.; Hemberger, M.; Pfarrer, C.; Cross, J.C. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev. Biol. 2002, 250, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Kapranos, N.C.; Kotronias, D.C. Detection of herpes simplex virus in first trimester pregnancy loss using molecular techniques. In Vivo 2009, 23, 839–842. [Google Scholar] [PubMed]
- Oliveira, G.M.; Pascoal-Xavier, M.A.; Moreira, D.R.; Guimarães, V.S.; Aguiar, R.; Miranda, D.M.; Romanelli, R.M.C. Detection of cytomegalovirus, herpes virus simplex, and parvovirus b19 in spontaneous abortion placentas. J. Matern. Fetal Neonatal Med. 2019, 32, 768–775. [Google Scholar] [CrossRef]
- Cross, J.C.; Hemberger, M.; Lu, Y.; Nozaki, T.; Whiteley, K.; Masutani, M.; Adamson, S.L. Trophoblast functions, angiogenesis and remodeling of the maternal vasculature in the placenta. Mol. Cell. Endocrinol 2002, 187, 207–212. [Google Scholar] [CrossRef]
- Simmons, D.G.; Cross, J.C. Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev. Biol. 2005, 284, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Redhead, M.L.; Portilho, N.A.; Felker, A.M.; Mohammad, S.; Mara, D.L.; Croy, B.A. The Transcription Factor NFIL3 Is Essential for Normal Placental and Embryonic Development but Not for Uterine Natural Killer (UNK) Cell Differentiation in Mice. Biol. Reprod. 2016, 94, 101. [Google Scholar] [CrossRef] [Green Version]
- Kaushic, C. HIV-1 infection in the female reproductive tract: Role of interactions between HIV-1 and genital epithelial cells. Am. J. Reprod. Immunol. 2011, 65, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Wessels, J.M.; Felker, A.M.; Dupont, H.A.; Kaushic, C. The relationship between sex hormones, the vaginal microbiome and immunity in HIV-1 susceptibility in women. Dis. Model. Mech. 2018, 11, dmm035147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wira, C.R.; Rodriguez-Garcia, M.; Patel, M.V. The role of sex hormones in immune protection of the female reproductive tract. Nat. Rev. Immunol. 2015, 15, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Racicot, K.; Aldo, P.; El-Guindy, A.; Kwon, J.-Y.; Romero, R.; Mor, G. Cutting Edge: Fetal/Placental Type I IFN Can Affect Maternal Survival and Fetal Viral Load during Viral Infection. J. Immunol. 2017, 198, 3029–3032. [Google Scholar] [CrossRef] [Green Version]
- Thomson, K.A.; Hughes, J.; Baeten, J.M.; John-Stewart, G.; Celum, C.; Cohen, C.R.; Ngure, K.; Kiarie, J.; Mugo, N.; Heffron, R. Increased Risk of HIV Acquisition Among Women Throughout Pregnancy and During the Postpartum Period: A Prospective Per-Coital-Act Analysis Among Women With HIV-Infected Partners. J. Infect. Dis. 2018, 218, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, L.D.; Ellington, S.; Strid, P.; Galang, R.R.; Oduyebo, T.; Tong, V.T.; Woodworth, K.R.; Nahabedian, J.F., 3rd; Azziz-Baumgartner, E.; Gilboa, S.M.; et al. Update: Characteristics of Symptomatic Women of Reproductive Age with Laboratory-Confirmed SARS-CoV-2 Infection by Pregnancy Status—United States, January 22-October 3, 2020. Morb. Mortal. Wkl. Rep. 2020, 69, 1641–1647. [Google Scholar] [CrossRef]
- Young, E.J.; Gomez, C.I. Enhancement of herpesvirus type 2 infection in pregnant mice. Proc. Soc. Exp. Biol. Med. 1979, 160, 416–420. [Google Scholar] [CrossRef]
- Bujko, M.; Sulović, V.; Zivanović, V.; Lako, B.; Dotlić, R. Effect of progesterone and pregnancy on the replication of herpes simplex virus type 2 in vivo. Clin. Exp. Obstet. Gynecol. 1988, 15, 34–37. [Google Scholar]
- McDonagh, S.; Maidji, E.; Ma, W.; Chang, H.-T.; Fisher, S.; Pereira, L. Viral and bacterial pathogens at the maternal-fetal interface. J. Infect. Dis. 2004, 190, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Chen, Y.; Fang, Z.; Chen, Q.; Chen, L.; Han, Q.; Yan, J. Effects of cytomegalovirus infection on extravillous trophoblast cells invasion and immune function of NK cells at the maternal-fetal interface. J. Cell. Mol. Med. 2020, 24, 11170–11176. [Google Scholar] [CrossRef]
- Kingdom, J.; Huppertz, B.; Seaward, G.; Kaufmann, P. Development of the placental villous tree and its consequences for fetal growth. Eur. J. Obs. Gynecol. Reprod. Biol. 2000, 92, 35–43. [Google Scholar] [CrossRef]
- Kawakami, T.; Yoshimi, M.; Kadota, Y.; Inoue, M.; Sato, M.; Suzuki, S. Prolonged endoplasmic reticulum stress alters placental morphology and causes low birth weight. Toxicol. Appl. Pharmacol. 2014, 275, 134–144. [Google Scholar] [CrossRef]
- Perez-Garcia, V.; Fineberg, E.; Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J.; et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Pásztor, N.; Sikovanyecz, J.; Keresztúri, A.; Kozinszky, Z.; Németh, G. Evaluation of the relation between placental weight and placental weight to foetal weight ratio and the causes of stillbirth: A retrospective comparative study. J. Obstet. Gynaecol. 2018, 38, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Syridou, G.; Spanakis, N.; Konstantinidou, A.; Piperaki, E.-T.; Kafetzis, D.; Patsouris, E.; Antsaklis, A.; Tsakris, A. Detection of cytomegalovirus, parvovirus B19 and herpes simplex viruses in cases of intrauterine fetal death: Association with pathological findings. J. Med. Virol. 2008, 80, 1776–1782. [Google Scholar] [CrossRef]
- Sifakis, S.; Koumantakis, E.; Koffa, M.; Ergazaki, M.; Spandidos, D.A. Detection of herpes simplex virus (HSV) in aborted material using the polymerase chain reaction technique. Gynecol. Obstet. Investig. 1998, 45, 109–115. [Google Scholar] [CrossRef]
- Page, J.M.; Bardsley, T.; Thorsten, V.; Allshouse, A.A.; Varner, M.W.; Debbink, M.P.; Dudley, D.J.; Saade, G.R.; Goldenberg, R.L.; Stoll, B.; et al. Stillbirth Associated With Infection in a Diverse, U.S. Obste.t Gynecol. 2019, 134, 1187–1196. [Google Scholar] [CrossRef]
- Nasyrov, R.A.; Sidorova, N.A.; Melnikova, V.F.; Fedotova, E.P. Morphological and Immunohistochemical Features of Placental Damage in Cases of Perinatal Death: Institutional Experience with Emphasis on Viral Etiology. Ann. Clin. Lab. Sci. 2020, 50, 754–760. [Google Scholar]
- Smith, A.E.; McKenney, A.; Rabinowitz, L.; Das, A. Diagnosis of Neonatal Herpes Simplex Infection from the Placenta. Case Rep. Pediatr. 2020, 2020, 8898612. [Google Scholar] [CrossRef]
- Greer, I.A.; Lyall, F.; Perera, T.; Boswell, F.; Macara, L.M. Increased concentrations of cytokines interleukin-6 and interleukin-1 receptor antagonist in plasma of women with preeclampsia: A mechanism for endothelial dysfunction? Obstet. Gynecol. 1994, 84, 937–940. [Google Scholar]
- Hirsch, E.; Blanchard, R.; Mehta, S.P. Differential fetal and maternal contributions to the cytokine milieu in a murine model of infection-induced preterm birth. Am. J. Obstet. Gynecol. 1999, 180, 429–434. [Google Scholar] [CrossRef]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Parisi, F.; Milazzo, R.; Savasi, V.M.; Cetin, I. Maternal Low-Grade Chronic Inflammation and Intrauterine Programming of Health and Disease. Int. J. Mol. Sci. 2021, 22, 1732. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, O.V.; Lalayan, D.V.; Sel’kov, S.A. Spontaneous and LPS-induced secretion of cytokines by villous chorion tissue. Bull. Exp. Biol. Med. 2006, 141, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Paulesu, L.; Jantra, S.; Ietta, F.; Brizzi, R.; Bigliardi, E. Interleukin-1 in reproductive strategies. Evol. Dev. 2008, 10, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Paulesu, L.; King, A.; Loke, Y.W.; Cintorino, M.; Bellizzi, E.; Boraschi, D. Immunohistochemical localization of IL-1 alpha and IL-1 beta in normal human placenta. Lymphokine Cytokine Res. 1991, 10, 443–448. [Google Scholar]
- Hu, X.L.; Yang, Y.; Hunt, J.S. Differential distribution of interleukin-1 alpha and interleukin-1 beta proteins in human placentas. J. Reprod. Immunol. 1992, 22, 257–268. [Google Scholar] [CrossRef]
- De, M.; Sanford, T.H.; Wood, G.W. Detection of interleukin-1, interleukin-6, and tumor necrosis factor-alpha in the uterus during the second half of pregnancy in the mouse. Endocrinology 1992, 131, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.R.; Devoto, L.; Campana, A.; Bischof, P. Effects of leptin, interleukin-1alpha, interleukin-6, and transforming growth factor-beta on markers of trophoblast invasive phenotype: Integrins and metalloproteinases. Endocrine 2001, 15, 157–164. [Google Scholar] [CrossRef]
- Cotechini, T.; Graham, C.H. Aberrant maternal inflammation as a cause of pregnancy complications: A potential therapeutic target? Placenta 2015, 36, 960–966. [Google Scholar] [CrossRef]
- Eloundou, S.N.; Lee, J.; Wu, D.; Lei, J.; Feller, M.C.; Ozen, M.; Zhu, Y.; Hwang, M.; Jia, B.; Xie, H.; et al. Placental malperfusion in response to intrauterine inflammation and its connection to fetal sequelae. PLoS ONE 2019, 14, e0214951. [Google Scholar] [CrossRef] [PubMed]
- Cotechini, T.; Komisarenko, M.; Sperou, A.; Macdonald-Goodfellow, S.; Adams, M.A.; Graham, C.H. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J. Exp. Med. 2014, 211, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Shin, N.E.; Na, Q.; Dong, J.; Chudnovets, A.; Li, S.; Novak, C.M.; McLane, M.W.; Lei, J.; Burd, I. Exposure to systemic and intrauterine inflammation leads to decreased pup survival via different placental mechanisms. J. Reprod. Immunol. 2019, 133, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Pathology Score | No. of Mice | No. of Days | Cumulative Pathology | Avg. Pathology/Mouse (avg. ± SEM) |
|---|---|---|---|---|---|
| Diestrus | |||||
| 102 pfu/mL | 0 | 6 | 8 | 0 | 0.7 ± 0.62 |
| 5 | 1 | 1 | 5 | ||
| 103 pfu/mL | 0 | 5 | 8 | 0 | 0.9 ± 0.59 |
| 1 | 1 | 2 | 2 | ||
| 2 | 1 | 2 | 4 | ||
| 104 pfu/mL | 0 | 3 | 8 | 0 | 1.5 ± 0.72 |
| 1 | 1 | 3 | 3 | ||
| 2 | 1 | 1 | 2 | ||
| 4 | 1 | 1 | 4 | ||
| 105 pfu/mL | 0 | 1 | 8 | 0 | 5.0 ± 0.98 |
| 2 | 1 | 3 | 6 | ||
| 3 | 1 | 2 | 6 | ||
| 4 | 2 | 1 | 8 | ||
| 5 | 2 | 1 | 10 | ||
| 5 | 1 | 2 | 10 | ||
| Pregnant | |||||
| 102 pfu/mL | 1 | 1 | 2 | 2 | 2.8 ± 0.48 b |
| 2 | 1 | 1 | 2 | ||
| 2 | 1 | 2 | 4 | ||
| 3 | 1 | 1 | 3 | ||
| 103 pfu/mL | 1 | 1 | 2 | 2 | 4.2 ± 1.0 b |
| 3 | 1 | 1 | 3 | ||
| 4 | 2 | 1 | 8 | ||
| 4 | 1 | 2 | 8 | ||
| 104 pfu/mL | 4 | 1 | 1 | 4 | 6.7 ± 1.3b |
| 4 | 2 | 2 | 16 | ||
| 105 pfu/mL | 4 | 1 | 1 | 4 | 8.2 ± 1.7 |
| 4 | 1 | 3 | 12 | ||
| 5 | 4 | 1 | 20 | ||
| 5 | 2 | 3 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felker, A.M.; Nguyen, P.; Kaushic, C. Primary HSV-2 Infection in Early Pregnancy Results in Transplacental Viral Transmission and Dose-Dependent Adverse Pregnancy Outcomes in a Novel Mouse Model. Viruses 2021, 13, 1929. https://doi.org/10.3390/v13101929
Felker AM, Nguyen P, Kaushic C. Primary HSV-2 Infection in Early Pregnancy Results in Transplacental Viral Transmission and Dose-Dependent Adverse Pregnancy Outcomes in a Novel Mouse Model. Viruses. 2021; 13(10):1929. https://doi.org/10.3390/v13101929
Chicago/Turabian StyleFelker, Allison M., Philip Nguyen, and Charu Kaushic. 2021. "Primary HSV-2 Infection in Early Pregnancy Results in Transplacental Viral Transmission and Dose-Dependent Adverse Pregnancy Outcomes in a Novel Mouse Model" Viruses 13, no. 10: 1929. https://doi.org/10.3390/v13101929
APA StyleFelker, A. M., Nguyen, P., & Kaushic, C. (2021). Primary HSV-2 Infection in Early Pregnancy Results in Transplacental Viral Transmission and Dose-Dependent Adverse Pregnancy Outcomes in a Novel Mouse Model. Viruses, 13(10), 1929. https://doi.org/10.3390/v13101929