
Coxsackievirus A2 Leads to Heart Injury in a Neonatal Mouse Model


 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Mice and Infection Model
2.3. Immunofluorescent Staining
2.4. Quantitative RT-PCR (qRT-PCR)
2.5. ELISA
2.6. Detection of Apoptosis
2.7. Tissue Histopathology
2.8. Examination of Cardiac Enzymes
2.9. Western Blot (WB)
2.10. Antibodies
2.11. Statistical Analysis
3. Results
3.1. CVA2 Infection Led to Heart Injury in a Mouse Model
3.2. Alterations of Cardiac Enzymes in CVA2-Induced Acute Heart Injury
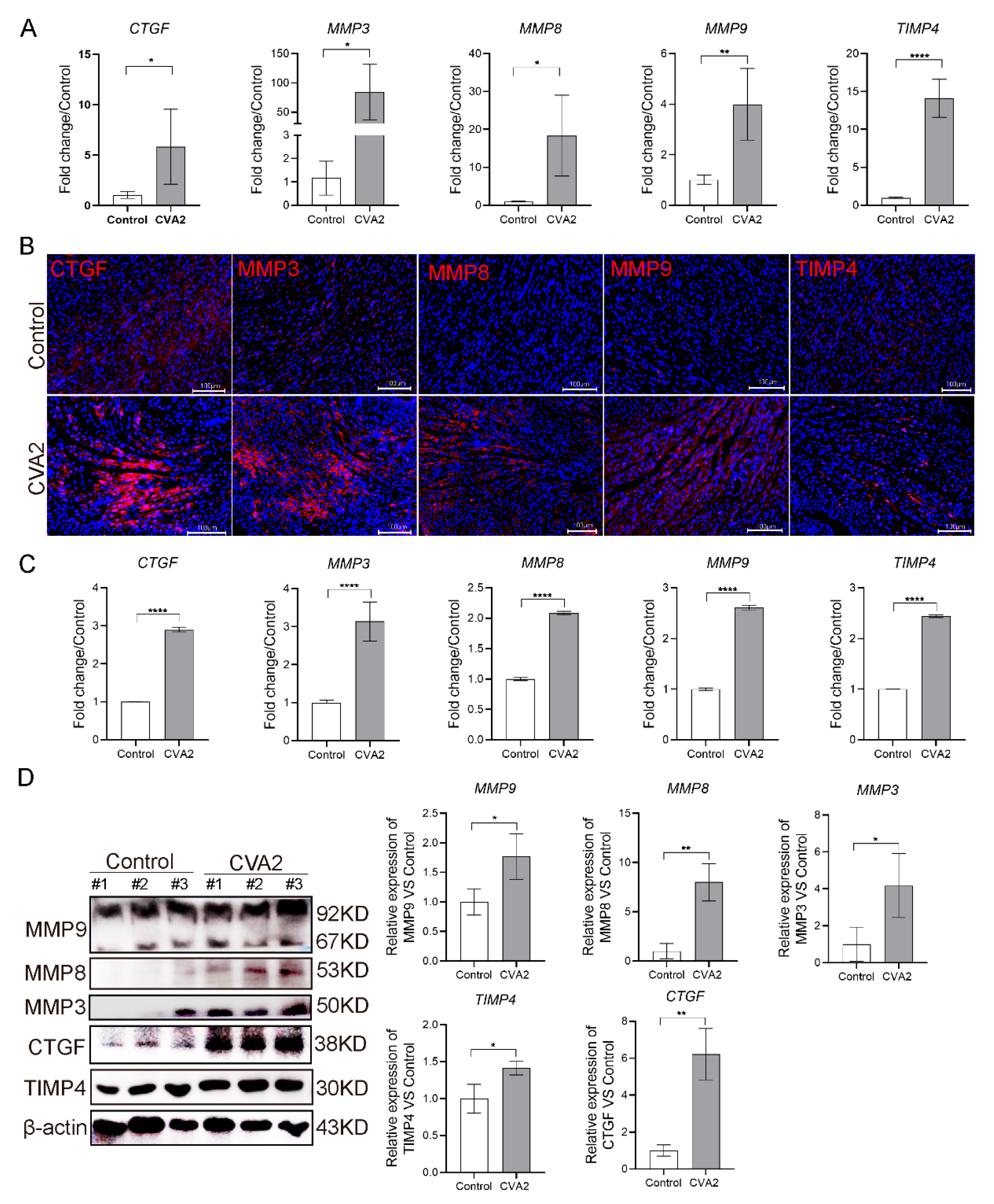
3.3. CVA2 Infection Led to the Disruption of Cell-Matrix Interactions in Heart Tissues
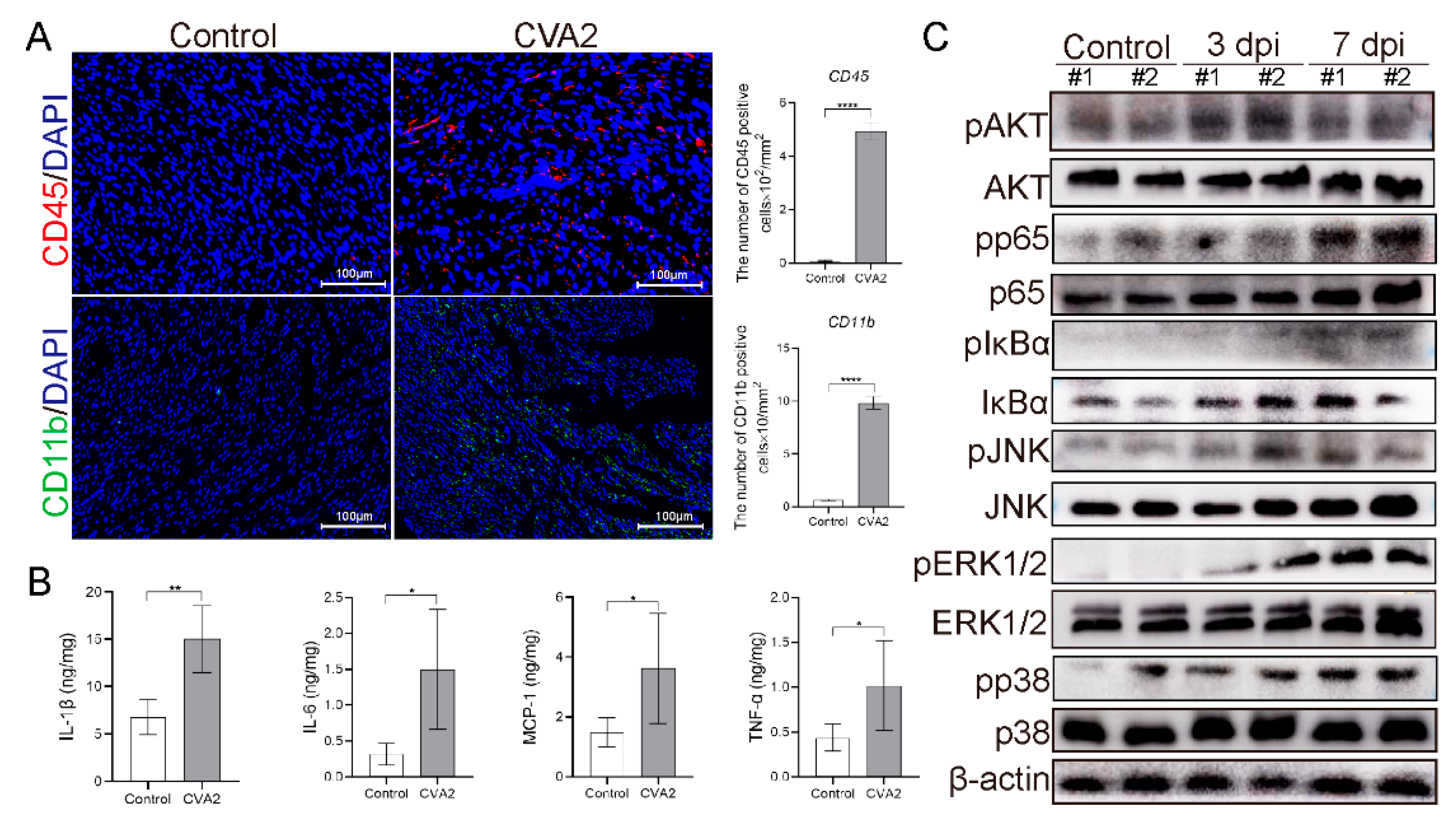
3.4. Leukocyte Infiltration and Proinflammatory Cytokines Expression in Heart Tissues after CVA2 Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.; Xu, S.J.; Yao, X.J.; Yang, H.; Zhang, H.L.; Meng, J.; Zeng, H.R.; Huang, X.H.; Zhang, R.L.; He, Y.Q. Molecular Epidemiology of Enteroviruses Associated with Severe Hand, Foot and Mouth Disease in Shenzhen, China, 2014–2018. Arch. Virol. 2020, 165, 2213–2227. [Google Scholar] [CrossRef]
- Yang, Q.; Gu, X.; Zhang, Y.; Wei, H.; Li, Q.; Fan, H.; Xu, Y.; Li, J.; Tan, Z.; Song, Y.; et al. Persistent Circulation of Genotype D Coxsackievirus A2 in Mainland of China since 2008. PLoS ONE 2018, 13, e0204359. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, H.H.; Zhang, Q.; Liu, Y.J.; Tao, R.; Cheng, Y.P.; Shu, Q.; Shang, S.Q. Large Outbreak of Herpangina in Children Caused by Enterovirus in Summer of 2015 in Hangzhou, China. Sci. Rep. 2016, 6, 35388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.Y.; Chiang, P.S.; Luo, S.T.; Lin, T.Y.; Tsao, K.C.; Lee, M.S. A Molecular Approach Applied to Enteroviruses Surveillance in Northern Taiwan, 2008-2012. PLoS ONE 2016, 11, e0167532. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, Y.; Yan, D.; Zhu, S.; Wang, D.; Ji, T.; Li, X.; Song, Y.; Gu, X.; Xu, W. Two Genotypes of Coxsackievirus A2 Associated with Hand, Foot, and Mouth Disease Circulating in China since 2008. PLoS ONE 2016, 11, e0169021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, K.L.; Wei, S.H.; Fan, H.C.; Chou, Y.K.; Yang, J.Y. Outbreak of Recombinant Coxsackievirus A2 Infection and Polio-Like Paralysis of Children, Taiwan, 2014. Pediatr. Neonatol. 2019, 60, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Yen, T.Y.; Huang, Y.P.; Hsu, Y.L.; Chang, Y.T.; Lin, H.C.; Wu, H.S.; Hwang, K.P. A Case of Recombinant Coxsackievirus A2 Infection with Neurological Complications in Taiwan. J. Microbiol. Immunol. Infect. 2017, 50, 928–930. [Google Scholar] [CrossRef]
- Chansaenroj, J.; Auphimai, C.; Puenpa, J.; Mauleekoonphairoj, J.; Wanlapakorn, N.; Vuthitanachot, V.; Vongpunsawad, S.; Poovorawan, Y. High Prevalence of Coxsackievirus A2 in Children with Herpangina in Thailand in 2015. Virusdisease 2017, 28, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Molet, L.; Saloum, K.; Marque-Juillet, S.; Garbarg-Chenon, A.; Henquell, C.; Schuffenecker, I.; Peigue-Lafeuille, H.; Rozenberg, F.; Mirand, A. Enterovirus Infections in Hospitals of Ile De France Region over 2013. J. Clin. Virol. 2016, 74, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Hoa-Tran, T.N.; Nguyen, A.T.; Dao, A.T.H.; Kataoka, C.; Ta, H.T.T.; Nguyen, H.T.V.; Takemura, T.; Nguyen, T.T.T.; Vu, H.M.; Nguyen, T.T.H.; et al. Genetic Characterization of Vp1 of Coxsackieviruses A2, A4, and A10 Associated with Hand, Foot, and Mouth Disease in Vietnam in 2012-2017: Endemic Circulation and Emergence of New Hfmd-Causing Lineages. Arch. Virol. 2020, 165, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.P., Jr.; Oliveira, M.L.A.; Burlandy, F.M.; Machado, R.S.; Oliveira, S.S.; Tavares, F.N.; Gomes-Neto, F.; da Costa, E.V.; da Silva, E.E. Molecular Characterization and Epidemiological Aspects of Non-Polio Enteroviruses Isolated from Acute Flaccid Paralysis in Brazil: A Historical Series (2005–2017). Emerg. Microbes Infect. 2020, 9, 2536–2546. [Google Scholar] [CrossRef]
- Park, K.; Lee, B.; Baek, K.; Cheon, D.; Yeo, S.; Park, J.; Soh, J.; Cheon, H.; Yoon, K.; Choi, Y. Enteroviruses Isolated from Herpangina and Hand-Foot-and-Mouth Disease in Korean Children. Virol. J. 2012, 9, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Choi, S.S.; Oh, S.A.; Kim, C.K.; Cho, S.J.; Lee, J.H.; Ryu, S.H.; Pak, S.H.; Jung, S.K.; Lee, J.I.; et al. Detection and Characterization of Enterovirus Associated with Herpangina and Hand, Foot, and Mouth Disease in Seoul, Korea. Clin. Lab. 2011, 57, 959–967. [Google Scholar] [PubMed]
- Chen, S.P.; Huang, Y.C.; Li, W.C.; Chiu, C.H.; Huang, C.G.; Tsao, K.C.; Lin, T.Y. Comparison of Clinical Features between Coxsackievirus A2 and Enterovirus 71 During the Enterovirus Outbreak in Taiwan, 2008: A Children’s Hospital Experience. J. Microbiol. Immunol. Infect. 2010, 43, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Bendig, J.W.; O’Brien, P.S.; Muir, P.; Porter, H.J.; Caul, E.O. Enterovirus Sequences Resembling Coxsackievirus A2 Detected in Stool and Spleen from a Girl with Fatal Myocarditis. J. Med. Virol. 2001, 64, 482–486. [Google Scholar] [CrossRef]
- Ohara, N.; Kaneko, M.; Nishibori, T.; Sato, K.; Furukawa, T.; Koike, T.; Sone, H.; Kaneko, K.; Kamoi, K. Fulminant Type 1 Diabetes Mellitus Associated with Coxsackie Virus Type A2 Infection: A Case Report and Literature Review. Intern. Med. 2016, 55, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, C.C.; Lau, S.K.; Woo, P.C.; Wong, S.S.; Tsang, T.H.; Lo, J.Y.; Lam, W.K.; Tsang, C.C.; Chan, K.H.; Yuen, K.Y. Recombinant Coxsackievirus A2 and Deaths of Children, Hong Kong, 2012. Emerg. Infect. Dis. 2013, 19, 1285–1288. [Google Scholar] [CrossRef]
- Heymans, S.; Eriksson, U.; Lehtonen, J.; Cooper, L.T., Jr. The Quest for New Approaches In myocarditis and Inflammatory cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2348–2364. [Google Scholar] [CrossRef]
- Freund, M.W.; Kleinveld, G.; Krediet, T.G.; van Loon, A.M.; Verboon-Maciolek, M.A. Prognosis for Neonates with Enterovirus Myocarditis. Arch. Dis. Child. Fetal Neonatal Ed. 2010, 95, F206–F212. [Google Scholar] [CrossRef]
- Cheung, C.; Marchant, D.; Walker, E.K.; Luo, Z.; Zhang, J.; Yanagawa, B.; Rahmani, M.; Cox, J.; Overall, C.; Senior, R.M.; et al. Ablation of Matrix Metalloproteinase-9 Increases Severity of Viral Myocarditis in Mice. Circulation 2008, 117, 1574–1582. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Pauschinger, M.; De Palma, A.; Kallwellis-Opara, A.; Rutschow, S.; Swinnen, M.; Vanhoutte, D.; Gao, F.; Torpai, R.; Baker, A.H.; et al. Inhibition of Urokinase-Type Plasminogen Activator or Matrix Metalloproteinases Prevents Cardiac Injury and Dysfunction During Viral Myocarditis. Circulation 2006, 114, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and Function of Matrix Metalloproteinases and Timps. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauschinger, M.; Chandrasekharan, K.; Schultheiss, H.P. Myocardial Remodeling in Viral Heart Disease: Possible Interactions between Inflammatory Mediators and Mmp-Timp System. Heart Fail. Rev. 2004, 9, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Lin, T.Y.; Chiang, P.S.; Li, W.C.; Luo, S.T.; Tsao, K.C.; Liou, G.Y.; Huang, M.L.; Hsia, S.H.; Huang, Y.C.; et al. An Investigation of Epidemic Enterovirus 71 Infection in Taiwan, 2008: Clinical, Virologic, and Serologic Features. Pediatr. Infect. Dis. J. 2010, 29, 1030–1034. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Jiang, S.W.; Gu, W.Z.; Hu, A.R.; Lu, C.T.; Liang, X.Y.; Hu, Y.R.; Zhu, D.D.; Xie, L. Clinicopathologic Features and Molecular Analysis of Enterovirus 71 Infection: Report of an Autopsy Case from the Epidemic of Hand, Foot and Mouth Disease in China. Pathol. Int. 2012, 62, 565–570. [Google Scholar] [CrossRef]
- Chang, C.S.; Liao, C.C.; Liou, A.T.; Chang, Y.S.; Chang, Y.T.; Tzeng, B.H.; Chen, C.C.; Shih, C. Enterovirus 71 Targets the Cardiopulmonary System in a Robust Oral Infection Mouse Model. Sci. Rep. 2019, 9, 11108. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.P.; Miao, Z.P.; Xu, F.; Lu, H.J.; Sun, Y.S.; Xia, Y.; Chen, C.; Yang, Z.N.; Xia, S.C.; Jiang, J.M.; et al. An Adult Gerbil Model for Evaluating Potential Coxsackievirus A16 Vaccine Candidates. Vaccine 2019, 37, 5341–5349. [Google Scholar] [CrossRef]
- Muench, H.; Reed, L.J. A Simple Method of Estimating 50 Percent End-Points. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar]
- Ji, W.; Qin, L.; Tao, L.; Zhu, P.; Liang, R.; Zhou, G.; Chen, S.; Zhang, W.; Yang, H.; Duan, G.; et al. Neonatal Murine Model of Coxsackievirus A2 Infection for the Evaluation of Antiviral Therapeutics and Vaccination. Front. Microbiol. 2021, 12, 1310. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, T.; Zhou, G.; Li, D.; Chen, S.; Zhang, W.; Li, X.; Zhang, R.; Yang, H.; Duan, G. Pathogenesis Study of Enterovirus 71 Using a Novel Human Scarb2 Knock-in Mouse Model. mSphere 2021, 6, e01048-20. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoades, R.E.; Tabor-Godwin, J.M.; Tsueng, G.; Feuer, R. Enterovirus Infections of the Central Nervous System. Virology 2011, 411, 288–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue Tropism, Pathology and Pathogenesis of Enterovirus Infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, B.M.; Chow, L.H.; Wilson, J.E.; Anderson, D.R.; Gulizia, J.M.; Gauntt, C.J.; Klingel, K.E.; Beisel, K.W.; Kandolf, R. Direct Myocardial Injury by Enterovirus: A Central Role in the Evolution of Murine Myocarditis. Clin. Immunol. Immunopathol. 1993, 68, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Badorff, C.; Lee, G.H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral Protease 2a Cleaves Dystrophin: Evidence of Cytoskeletal Disruption in an Acquired Cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [CrossRef]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current State of Knowledge on Aetiology, Diagnosis, Management, and Therapy of Myocarditis: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and Inflammatory Cardiomyopathy: Current Evidence and Future Directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Jan, S.L.; Lin, S.J.; Fu, Y.C.; Lin, M.C.; Chan, S.C.; Hwang, B. Plasma B-Type Natriuretic Peptide Study in Children with Severe Enterovirus 71 Infection: A Pilot Study. Int. J. Infect. Dis. 2013, 17, e1166–e1171. [Google Scholar] [CrossRef] [Green Version]
- Lauer, B.; Niederau, C.; Kühl, U.; Schannwell, M.; Pauschinger, M.; Strauer, B.E.; Schultheiss, H.P. Cardiac Troponin T in Patients with Clinically Suspected Myocarditis. J. Am. Coll Cardiol. 1997, 30, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.C.; Ladenson, J.H.; Mason, J.W.; Jaffe, A.S. Elevations of Cardiac Troponin I Associated with Myocarditis. Experimental and Clinical Correlates. Circulation 1997, 95, 163–168. [Google Scholar] [CrossRef]
- Becher, P.M.; Gotzhein, F.; Klingel, K.; Escher, F.; Blankenberg, S.; Westermann, D.; Lindner, D. Cardiac Function Remains Impaired Despite Reversible Cardiac Remodeling after Acute Experimental Viral Myocarditis. J. Immunol. Res. 2017, 2017, 6590609. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Savvatis, K.; Schultheiss, H.P.; Tschöpe, C. Immunomodulation and Matrix Metalloproteinases in Viral Myocarditis. J. Mol. Cell. Cardiol. 2010, 48, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 Replication and Pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef]
- Pauschinger, M.; Rutschow, S.; Chandrasekharan, K.; Westermann, D.; Weitz, A.; Peter Schwimmbeck, L.; Zeichhardt, H.; Poller, W.; Noutsias, M.; Li, J.; et al. Carvedilol Improves Left Ventricular Function in Murine Coxsackievirus-Induced Acute Myocarditis Association with Reduced Myocardial Interleukin-1beta and Mmp-8 Expression and a Modulated Immune Response. Eur. J. Heart Fail. 2005, 7, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.H.; Wang, Y.; Zhuang, J.X.; Han, X.Z.; Chen, Y.; Jin, Y.P.; Wang, Y.L.; Yu, Y.H.; Spires, J.P.; Song, G.J. Dynamic Changes in Myocardial Matrix Metalloproteinase Activity in Mice with Viral Myocarditis. Chin. Med. J. 2004, 117, 1195–1199. [Google Scholar] [PubMed]
- Lang, C.; Sauter, M.; Szalay, G.; Racchi, G.; Grassi, G.; Rainaldi, G.; Mercatanti, A.; Lang, F.; Kandolf, R.; Klingel, K. Connective Tissue Growth Factor: A Crucial Cytokine-Mediating Cardiac Fibrosis in Ongoing Enterovirus Myocarditis. J. Mol. Med. 2008, 86, 49–60. [Google Scholar] [CrossRef]
- Yun, S.H.; Shin, J.O.; Lim, B.K.; Kim, K.L.; Gil, C.O.; Kim, D.K.; Jeon, E.S. Change in the Cells That Express Connective Tissue Growth Factor in Acute Coxsackievirus-Induced Myocardial Fibrosis in Mouse. Virus Res. 2007, 126, 62–68. [Google Scholar] [CrossRef]
- Cheung, C.; Luo, H.; Yanagawa, B.; Leong, H.S.; Samarasekera, D.; Lai, J.C.; Suarez, A.; Zhang, J.; McManus, B.M. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Coxsackievirus-Induced Myocarditis. Cardiovasc. Pathol. 2006, 15, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.J.; Wang, W.; Suarez-Pinzon, W.L.; Sawicka, J.; Sawicki, G.; Schulz, R. Imbalance between Tissue Inhibitor of Metalloproteinase-4 and Matrix Metalloproteinases during Acute Myocardial [Correction of Myoctardial] Ischemia-Reperfusion Injury. Circulation 2003, 107, 2487–2492. [Google Scholar] [CrossRef]
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Proinflammatory Cytokines Regulate Tissue Inhibitors of Metalloproteinases and Disintegrin Metalloproteinase in Cardiac Cells. Cardiovasc. Res. 1999, 42, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Koskivirta, I.; Rahkonen, O.; Mäyränpää, M.; Pakkanen, S.; Husheem, M.; Sainio, A.; Hakovirta, H.; Laine, J.; Jokinen, E.; Vuorio, E.; et al. Tissue Inhibitor of Metalloproteinases 4 (Timp4) Is Involved in Inflammatory Processes of Human Cardiovascular Pathology. Histochem. Cell Biol. 2006, 126, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Liu, P.P.; Cooper, L.T., Jr. Myocarditis. Lancet 2012, 379, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of Biologically Active Il-1 Beta by Matrix Metalloproteinases: A Novel Caspase-1-Independent Pathway of Il-1 Beta Processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar] [PubMed]
- Nelissen, I.; Martens, E.; Van den Steen, P.E.; Proost, P.; Ronsse, I.; Opdenakker, G. Gelatinase B/Matrix Metalloproteinase-9 Cleaves Interferon-Beta and Is a Target for Immunotherapy. Brain 2003, 126, 1371–1381. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.D.; Marchant, D.J. Emerging Pharmacologic Targets and Treatments for Myocarditis. Pharmacol. Ther. 2016, 161, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral Myocarditis--Diagnosis, Treatment Options, and Current Controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Job, L.P. Cellular Immune Mechanisms in Coxsackievirus Group B, Type 3 Induced Myocarditis in Balb/C Mice. Adv. Exp. Med. Biol. 1983, 161, 491–508. [Google Scholar] [PubMed]
- Marchant, D.J.; Boyd, J.H.; Lin, D.C.; Granville, D.J.; Garmaroudi, F.S.; McManus, B.M. Inflammation in Myocardial Diseases. Circ. Res. 2012, 110, 126–144. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Xu, W.; Chu, Y.W.; Wang, Y.; Liu, Q.S.; Xiong, S.D. Coxsackievirus Group B Type 3 Infection Upregulates Expression of Monocyte Chemoattractant Protein 1 in Cardiac Myocytes, Which Leads to Enhanced Migration of Mononuclear Cells in Viral Myocarditis. J. Virol. 2004, 78, 12548–12556. [Google Scholar] [CrossRef] [Green Version]
- Cen, Z.; Li, Y.; Wei, B.; Wu, W.; Huang, Y.; Lu, J. The Role of B Cells in Regulation of Th Cell Differentiation in Coxsackievirus B3-Induced Acute Myocarditis. Inflammation 2021. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of Mrna Translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian Mapk Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in Nf-Κb Signaling Pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling Via the Nfκb System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhang, R.; Wu, W.; Duan, G. Antiviral and Inflammatory Cellular Signaling Associated with Enterovirus 71 Infection. Viruses 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5’->3’) | Reverse (5’->3’) | Product Lengths (bp) |
|---|---|---|---|
| CVA2-VP1 | TCAGTCCCATTCATGTCGCC | AATGCGTTGTTGGGGCATTG | 118 |
| CTGF | GGGAGAACTGTGTACGGAGC | AGTGCACACTCCGATCTTGC | 97 |
| MMP3 | TCTCAGGTTCCAGAGAGTTAGA | TGTCACTGGTACCAACCTATTC | 239 |
| MMP8 | TTGAGAAAGCTTTTCACGTCTG | CTTGAGACGAAAGCAATGTTGA | 97 |
| MMP9 | CAAAGACCTGAAAACCTCCAAC | GACTGCTTCTCTCCCATCATC | 105 |
| TIMP4 | GGCCGGAACTACCTTCTCACT | CACCCTCAGCAGCACATCTG | 75 |
| BNP | TGCTGGAGCTGATAAGAGAAAA | GAAGGACTCTTTTTGGGTGTTC | 96 |
| LDH | GGGAAAGTCTCTGGCTGATGAA | CTGTCACAGATATACTTTATCGGC | 140 |
| CK | AAACCCACAGACAAGCATAAGA | CTCTTCAGAGGGTAGTACTTGC | 233 |
| AST | ACAAGAACACACCAATCTACGT | ATAGGGCCGAATGTCCTTAAAA | 92 |
| CTNI | CACACGCCAAGAAAAAGTCTAA | GCATAAGTCCTGAAGCTCTTCA | 194 |
| Mouse β-actin | GTGCTATGTTGCTCTAGACTTCG | ATGCCACAGGATTCCATACC | 174 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, W.; Zhu, P.; Liang, R.; Zhang, L.; Zhang, Y.; Wang, Y.; Zhang, W.; Tao, L.; Chen, S.; Yang, H.; et al. Coxsackievirus A2 Leads to Heart Injury in a Neonatal Mouse Model. Viruses 2021, 13, 1588. https://doi.org/10.3390/v13081588
Ji W, Zhu P, Liang R, Zhang L, Zhang Y, Wang Y, Zhang W, Tao L, Chen S, Yang H, et al. Coxsackievirus A2 Leads to Heart Injury in a Neonatal Mouse Model. Viruses. 2021; 13(8):1588. https://doi.org/10.3390/v13081588
Chicago/Turabian StyleJi, Wangquan, Peiyu Zhu, Ruonan Liang, Liang Zhang, Yu Zhang, Yuexia Wang, Weiguo Zhang, Ling Tao, Shuaiyin Chen, Haiyan Yang, and et al. 2021. "Coxsackievirus A2 Leads to Heart Injury in a Neonatal Mouse Model" Viruses 13, no. 8: 1588. https://doi.org/10.3390/v13081588
APA StyleJi, W., Zhu, P., Liang, R., Zhang, L., Zhang, Y., Wang, Y., Zhang, W., Tao, L., Chen, S., Yang, H., Jin, Y., & Duan, G. (2021). Coxsackievirus A2 Leads to Heart Injury in a Neonatal Mouse Model. Viruses, 13(8), 1588. https://doi.org/10.3390/v13081588