
Efficient Transendothelial Migration of Latently HIV-1-Infected Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus and Infection
2.3. Cell Adhesion Assay
2.4. Transendothelial Migration (TEM) Assay
2.5. In-Vitro Reconstitution of the Lymphatic Endothelium
2.6. Flow Cytometry (FCM)
2.7. Statistical Analysis
3. Results
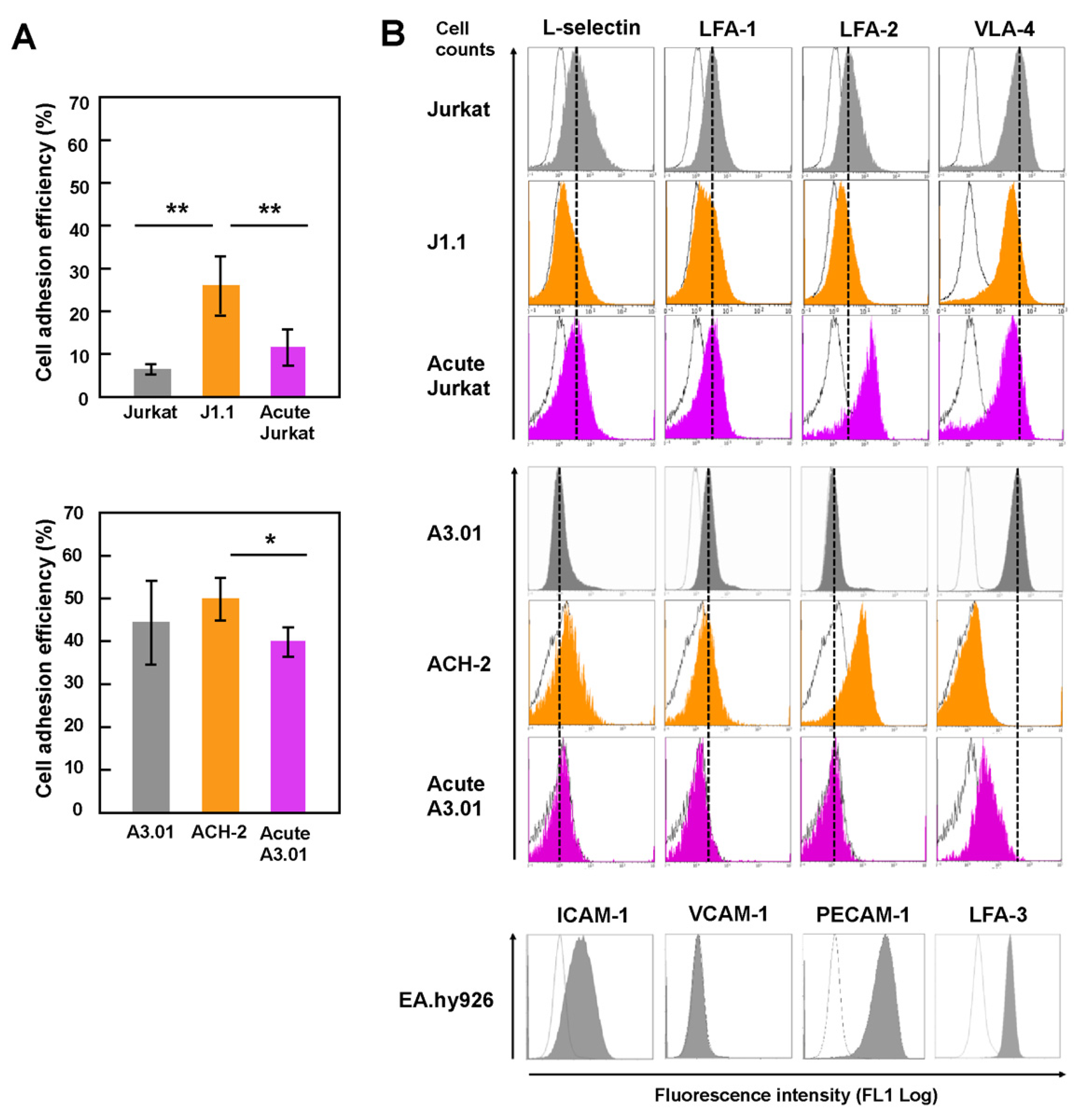
3.1. Latently HIV-1-Infected T Cells Efficiently Adhered to Endothelial Cells
3.2. Downregulation of the Expression of Surface Adhesion Molecules in Latently Infected T Cells
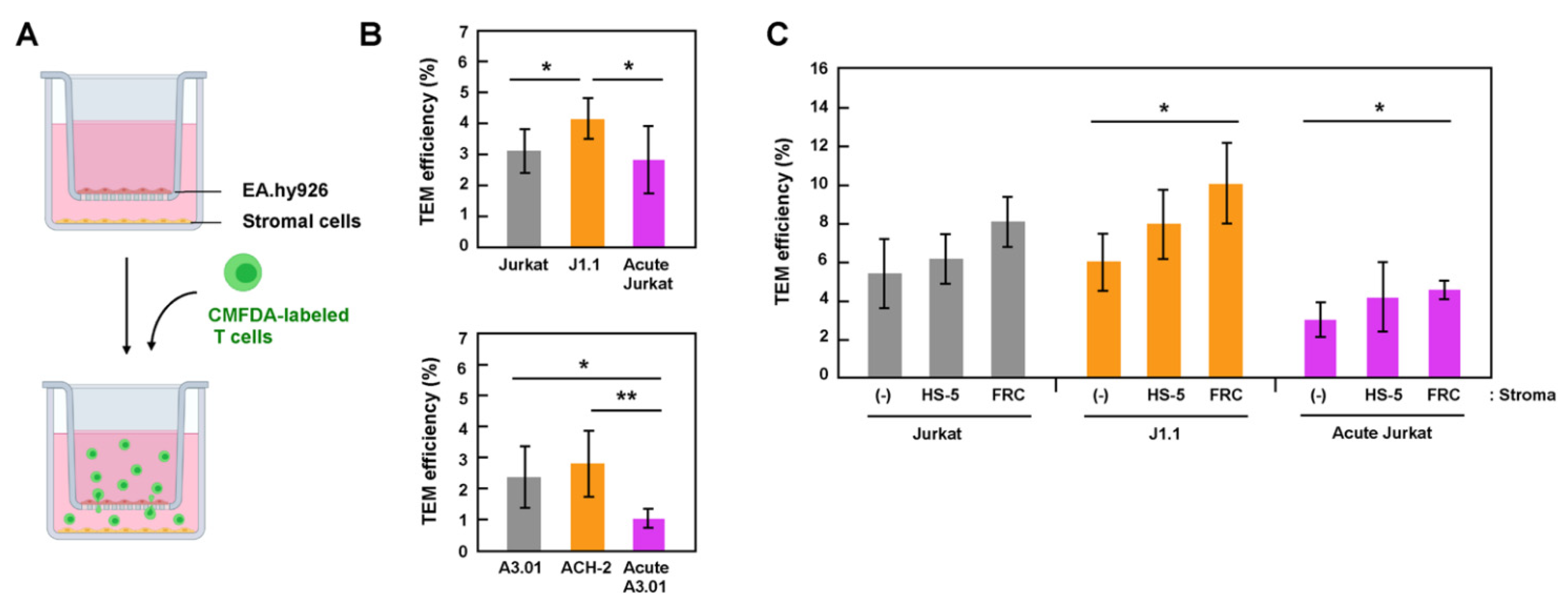
3.3. Latently Infected T Cells Efficiently Transmigrated across Endothelial Cells
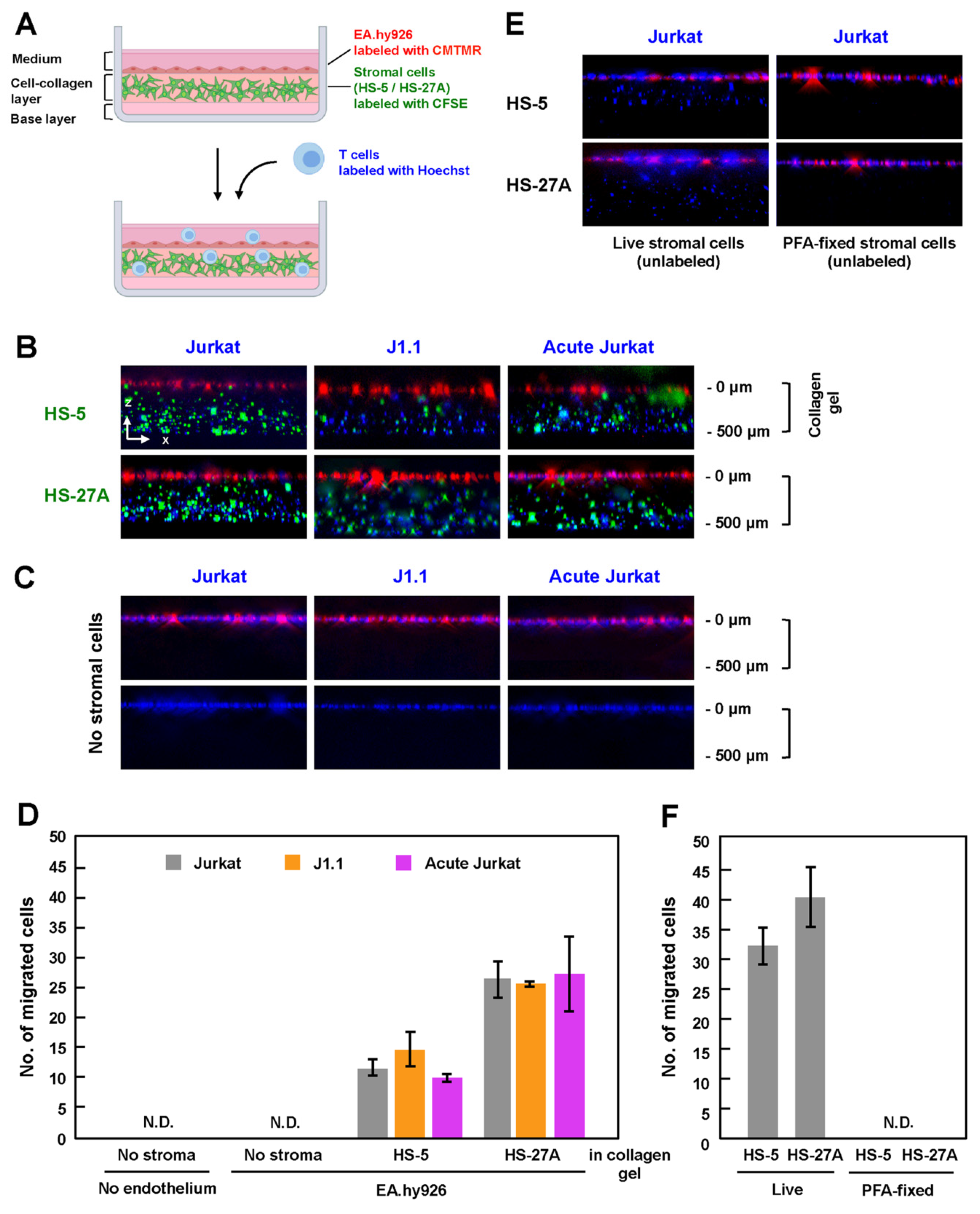
3.4. Efficient TEM in In-Vitro Reconstructed Lymphatic Endothelium Systems Required Live Stromal Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Gianella, S.; Sinclair, E.; Epling, L.; Li, Q.; Duan, L.; Choi, A.L.M.; Girling, V.; Ho, T.; Li, P.; et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J. Infect. Dis. 2010, 202, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.K.; Günthard, H.F.; Havlir, D.V.; Zhang, Z.Q.; Haase, A.T.; Ignacio, C.C.; Kwok, S.; Emini, E.; Richman, D.D. Reduction of HIV-1 in blood and lymph nodes following potent antiretroviral therapy and the virologic correlates of treatment failure. Proc. Natl. Acad. Sci. USA 1997, 94, 12574–12579. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Butcher, E.C.; Pircher, L.J. Lymphocyte Homing and Homeostasis. Science 1996, 272, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, E.; Tanaka, T.; Kanda, H.; Jin, S.; Tohya, K.; Otani, K.; Matsutani, T.; Matsumoto, M.; Ebisuno, Y.; Myoung, H.J.; et al. Nepmucin, a novel HEV sialomucin, mediates L-selectin-dependent lymphocyte rolling and promotes lymphocyte adhesion under flow. J. Exp. Med. 2006, 203, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, Z.; Shinder, V.; Klein, E.; Grabovsky, V.; Yeger, O.; Geron, E.; Montresor, A.; Bolomini-Vittori, M.; Feigelson, S.W.; Kirchhausen, T.; et al. Lymphocyte Crawling and Transendothelial Migration Require Chemokine Triggering of High-Affinity LFA-1 Integrin. Immunity 2009, 30, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Stanley, P.; Jones, K.; Svensson, L.; McDowall, A.; Hogg, N. The role of the integrin LFA-1 in T-lymphocyte migration. Immunol. Rev. 2007, 218, 135–146. [Google Scholar] [CrossRef]
- Dustin, M.L.; Sanders, M.E.; Shaw, S.; Springer, T.A. Purified lymphocyte function-associated antigen 3 binds to CD2 and mediates T lymphocyte adhesion. J. Exp. Med. 1987, 165, 677–692. [Google Scholar] [CrossRef]
- Muller, W.A. Platelet/Endothelial Cell Adhesion Molecule-1 (PECAM-1, CD31): The All-Purpose Adhesion Molecule. Trends Glycosci. Glycotechnol. 1994, 6, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; Von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.N.; Germain, R.N. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat. Rev. Immunol. 2009, 9, 618–629. [Google Scholar] [CrossRef]
- Turley, S.J.; Fletcher, A.L.; Elpek, K.G. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat. Rev. Immunol. 2010, 10, 813–825. [Google Scholar] [CrossRef]
- Saleh, S.; Solomon, A.; Wightman, F.; Xhilaga, M.; Cameron, P.U.; Lewin, S.R. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: A novel model of HIV-1 latency. Blood 2007, 110, 4161–4164. [Google Scholar] [CrossRef]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.P.; Harman, A.N.; et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2010, 107, 16934–16939. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2 + mobilization after CD3 stimulation. J. Immunol. 1991, 147, 3145–3148. [Google Scholar]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.S.; Folks, T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar] [PubMed]
- Lidington, E.A.; Moyes, D.L.; McCormack, A.M.; Rose, M.L. A comparison of primary endothelial cells and endothelial cell lines for studies of immune interactions. Transpl. Immunol. 1999, 7, 239–246. [Google Scholar] [CrossRef]
- Roecklein, B.; Torok-Storb, B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood 1995, 85, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.; Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Zentgraf, H.; Kräusslich, H.-G. Construction and Characterization of a Fluorescently Labeled Infectious Human Immunodeficiency Virus Type 1 Derivative. J. Virol. 2004, 78, 10803–10813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, W.; Chen, P.; Portillo, A.D.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of Human Immunodeficiency Virus Type 1 (HIV-1) Gag Localization and Oligomerization Monitored with Live Confocal Imaging of a Replication-Competent, Fluorescently Tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef] [Green Version]
- Tsunetsugu-Yokota, Y.; Yasuda, S.; Sugimoto, A.; Yagi, T.; Azuma, M.; Yagita, H.; Akagawa, K.; Takemori, T. Efficient virus transmission from dendritic cells to CD4+ T cells in response to antigen depends on close contact through adhesion molecules. Virology 1997, 239, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Schwartz, O.; Maréchal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2000, 2, 163–167. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-κB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, S.L.; Allen, T.D.; Winn, B. Lymphocyte migration into three-dimensional collagen matrices: A quantitative study. J. Cell Biol. 1983, 96, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-W.; He, J.J. HIV-1 Nef-mediated inhibition of T cell migration and its molecular determinants. J. Leukoc. Biol. 2009, 86, 1171–1178. [Google Scholar] [CrossRef]
- Vassena, L.; Giuliani, E.; Koppensteiner, H.; Bolduan, S.; Schindler, M.; Doria, M. HIV-1 Nef and Vpu Interfere with L-Selectin (CD62L) Cell Surface Expression To Inhibit Adhesion and Signaling in Infected CD4 + T Lymphocytes. J. Virol. 2015, 89, 5687–5700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.H.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.G.; Zack, J.A. Effect of Latent Human Immunodeficiency Virus Infection on Cell Surface Phenotype. J. Virol. 2002, 76, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Choe, E.Y.; Schoenberger, E.S.; Groopman, J.E.; Park, I.W. HIV Nef inhibits T cell migration. J. Biol. Chem. 2002, 277, 46079–46084. [Google Scholar] [CrossRef] [Green Version]
- Stolp, B.; Imle, A.; Coelho, F.M.; Hons, M.; Gorina, R.; Lyck, R.; Stein, J.V.; Fackler, O.T. HIV-1 Nef interferes with T-lymphocyte circulation through confined environments in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18541–18546. [Google Scholar] [CrossRef] [Green Version]
- Chandra, P.K.; Gerlach, S.L.; Wu, C.; Khurana, N.; Swientoniewski, L.T.; Abdel-Mageed, A.B.; Li, J.; Braun, S.E.; Mondal, D. Mesenchymal stem cells are attracted to latent HIV-1-infected cells and enable virus reactivation via a non-canonical PI3K-NFκB signaling pathway. Sci. Rep. 2018, 8, 14702. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanabe, R.; Morikawa, Y. Efficient Transendothelial Migration of Latently HIV-1-Infected Cells. Viruses 2021, 13, 1589. https://doi.org/10.3390/v13081589
Tanabe R, Morikawa Y. Efficient Transendothelial Migration of Latently HIV-1-Infected Cells. Viruses. 2021; 13(8):1589. https://doi.org/10.3390/v13081589
Chicago/Turabian StyleTanabe, Reou, and Yuko Morikawa. 2021. "Efficient Transendothelial Migration of Latently HIV-1-Infected Cells" Viruses 13, no. 8: 1589. https://doi.org/10.3390/v13081589
APA StyleTanabe, R., & Morikawa, Y. (2021). Efficient Transendothelial Migration of Latently HIV-1-Infected Cells. Viruses, 13(8), 1589. https://doi.org/10.3390/v13081589