
Apoptosis Enhances the Replication of Human Coronavirus OC43

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Amplification
2.3. Preparation of Virus-Infected Culture Supernatants
2.4. Plaque Assay
2.5. RT-qPCR
2.6. Cell Viability Assay
2.7. Apoptosis Assay
2.8. Cell Cycle Analysis
2.9. Western Blotting
2.10. Interferon-α Treatment
2.11. Inhibition of Apoptosis
2.12. Statistical Analysis
3. Results
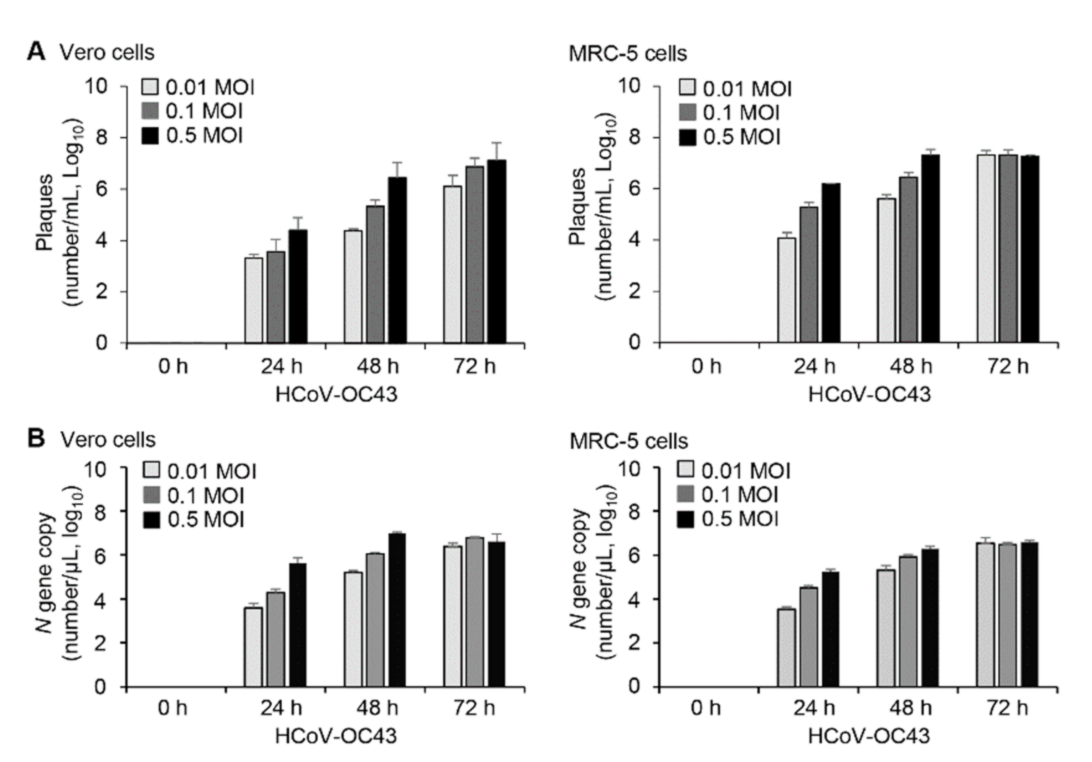
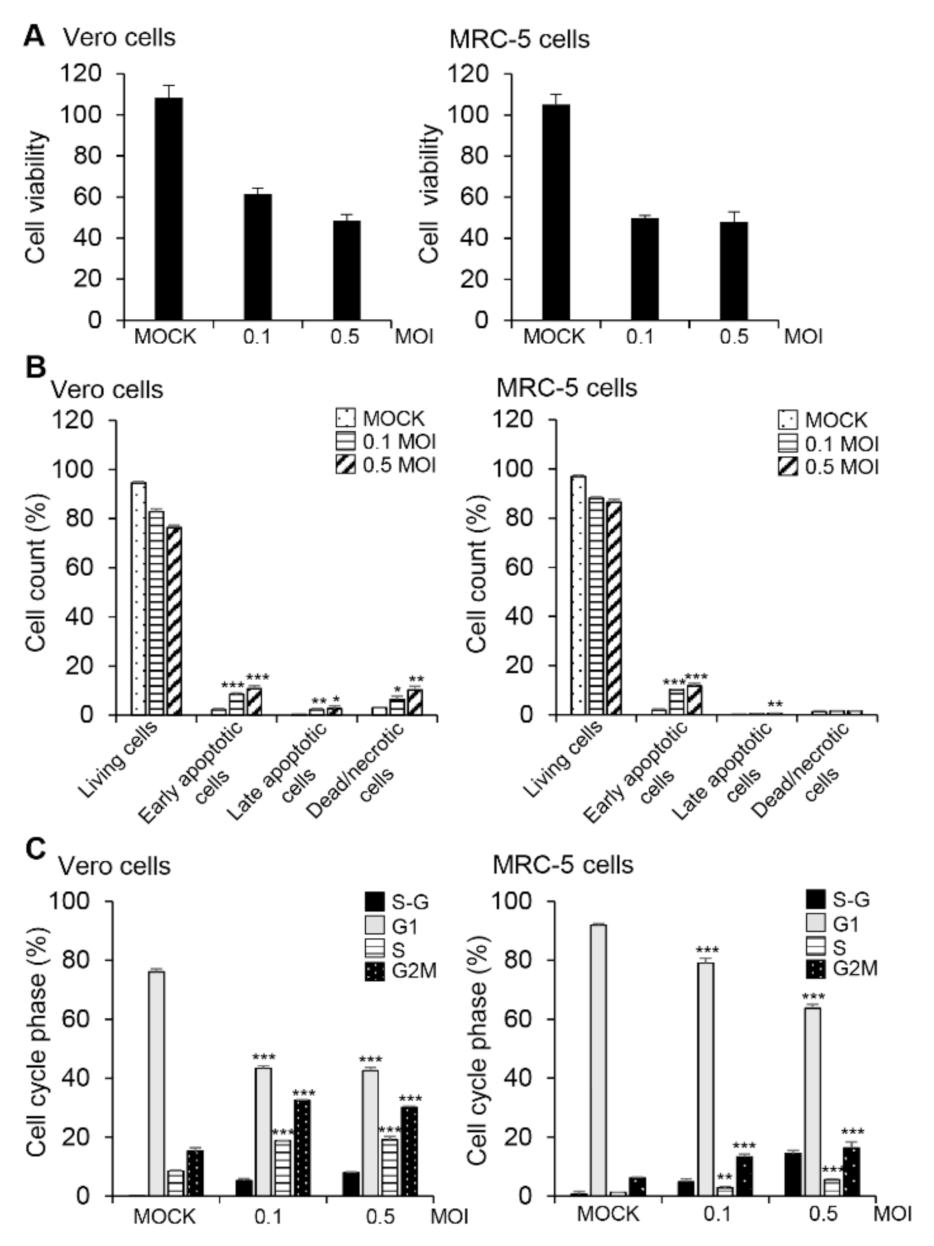
3.1. HCoV-OC43 Replication and Induction of Apoptosis in Vero and MRC-5 Cells
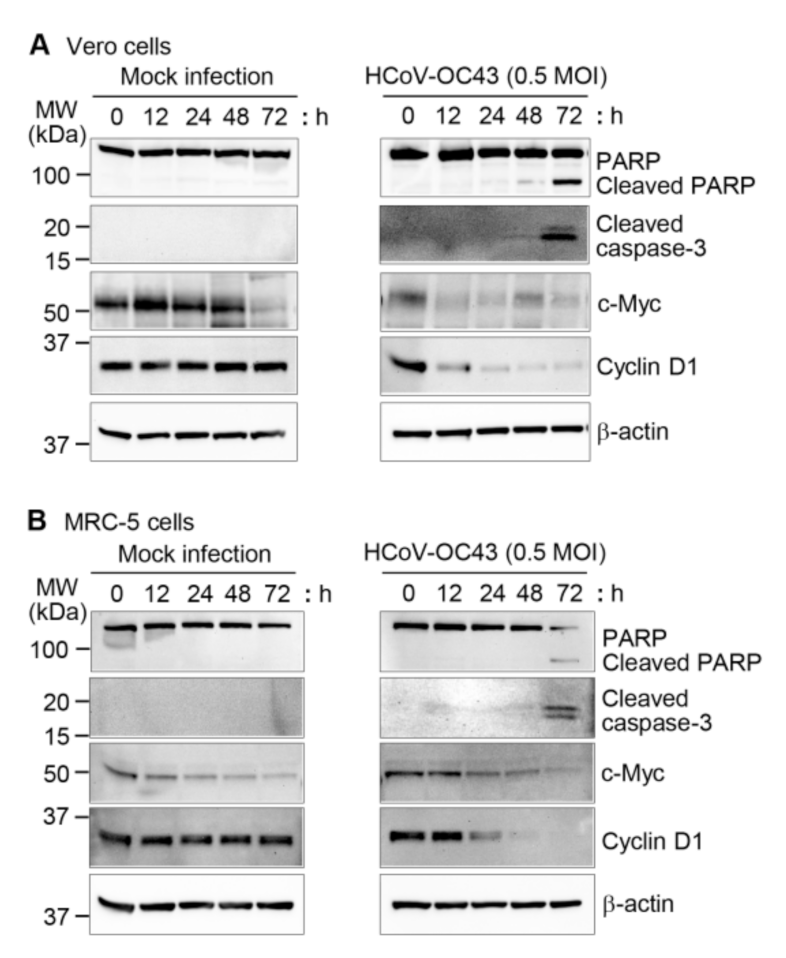
3.2. Effects of HCoV-OC43 Infection on Apoptotic and Cell Proliferation Marker Expression
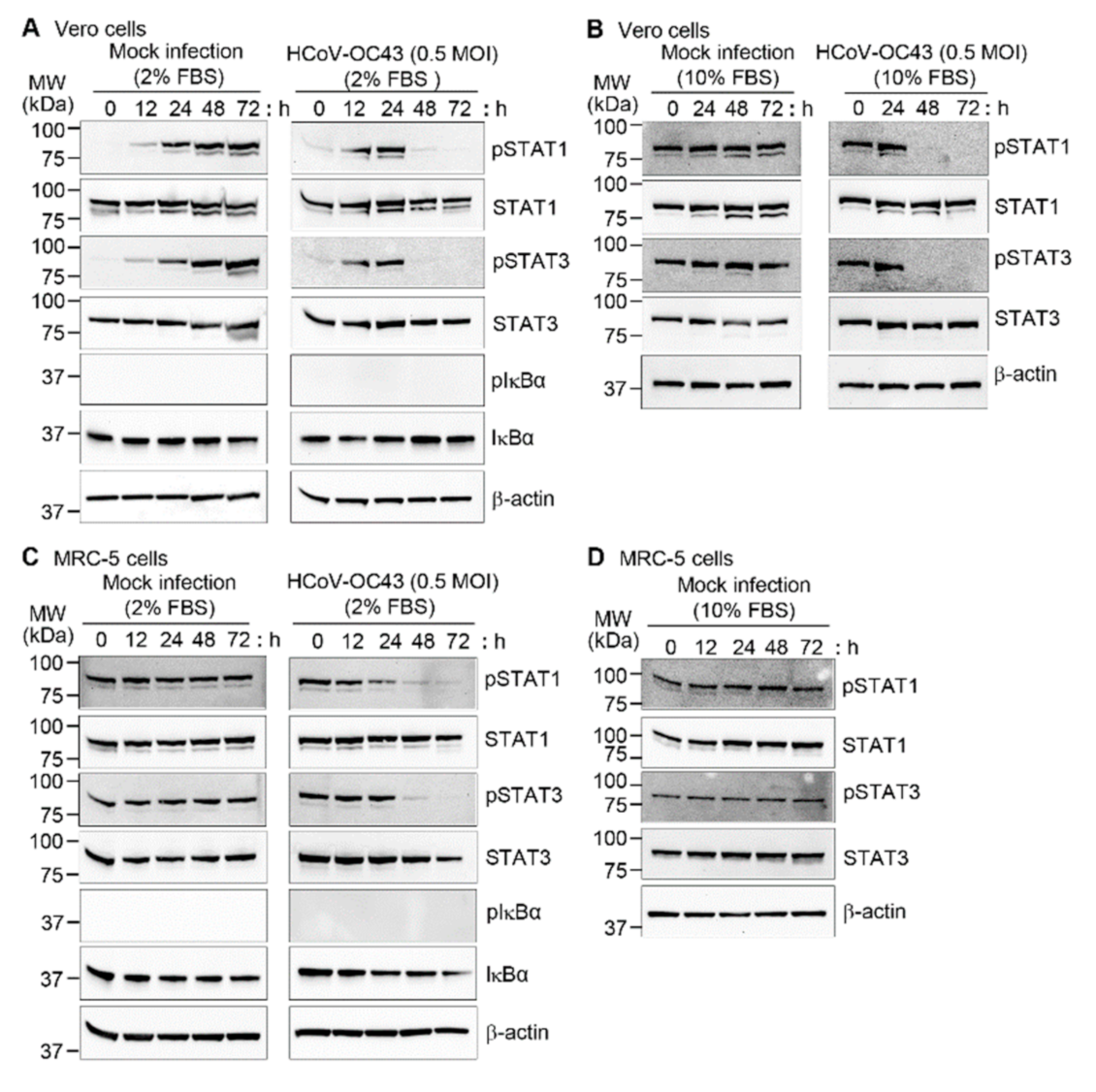
3.3. HCoV-OC43 Infection Decreased Phosphorylation of STAT1 and STAT3 in Vero and MRC-5 Cells
3.4. Suppression of Apoptosis and Reduction of the Viral Load by Caspase Inhibition
3.5. Activation of STAT 1 and STAT3 and Reduction of the Viral Load by Interferon-α
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCK-8 | Cell-counting kit-8 |
| CoVs | Coronaviruses |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | Dimethyl Sulfoxide |
| EMEM | Eagle’s Minimum Essential Medium |
| FACS | Fluorescence-activated cell sorting |
| FBS | Fetal bovine serum |
| HCoVs | Human coronaviruses |
| HCoV-OC43 | Human coronavirus OC43 |
| HRP | Horseradish peroxidase |
| IFN | Interferon |
| IκB | Inhibitory kappa B |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MOI | Multiplicity of infection |
| N | Nucleocapsid |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PARP | Poly-ADP ribose polymerase |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PI | Propidium iodide |
| RT-qPCR | Reverse transcriptase-quantitative PCR |
| S | Spike |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Zhao, X.; Ding, Y.; Du, J.; Fan, Y. 2020 update on human coronaviruses: One health, one world. Med. Nov. Technol. Devices 2020, 8, 100043. [Google Scholar] [CrossRef]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 2, pp. 428–440. [Google Scholar]
- Jacomy, H.; Fragoso, G.; Almazan, G.; Mushynski, W.E.; Talbot, P.J. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 2006, 349, 335–346. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon, Z.H.X.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19) Dashboard. 2021. Available online: https://www.who.int (accessed on 29 June 2021).
- Wenzel, R.P.; Hendley, J.O.; Davies, J.A.; Gwaltney, J.M., Jr. Coronavirus infections in military recruits. Three-year study with coronavirus strains OC43 and 229E. Am. Rev. Respir. Dis. 1974, 109, 621–624. [Google Scholar] [PubMed]
- Beidas, M.; Chehadeh, W. Effect of Human Coronavirus OC43 Structural and Accessory Proteins on the Transcriptional Activation of Antiviral Response Elements. Intervirology 2018, 61, 30–35. [Google Scholar] [CrossRef]
- Resta, S.; Luby, J.P.; Rosenfeld, C.R.; Siegel, J.D. Isolation and propagation of a human enteric coronavirus. Science 1985, 229, 978–981. [Google Scholar] [CrossRef]
- Riski, H.; Hovi, T. Coronavirus infections of man associated with diseases other than the common cold. J. Med. Virol. 1980, 6, 259–265. [Google Scholar] [CrossRef]
- McIntosh, K.; Chao, R.K.; Krause, H.E.; Wasil, R.; Mocega, H.E.; Mufson, M.A. Coronavirus infection in acute lower respiratory tract disease of infants. J. Infect. Dis. 1974, 130, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Morfopoulou, S.; Brown, J.R.; Davies, E.G.; Anderson, G.; Virasami, A.; Qasim, W.; Chong, W.K.; Hubank, M.; Plagnol, V.; Desforges, M.; et al. Human Coronavirus OC43 Associated with Fatal Encephalitis. N. Engl. J. Med. 2016, 375, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.A.; Collins, A.; Cohen, M.E.; Duffner, P.K.; Faden, H. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 2004, 113, e73–e76. [Google Scholar]
- Roulston, A.; Marcellus, R.C.; Branton, P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999, 53, 577–628. [Google Scholar] [CrossRef]
- Zhang, J.; Han, Y.; Shi, H.; Chen, J.; Zhang, X.; Wang, X.; Zhou, L.; Liu, J.; Zhang, J.; Ji, Z.; et al. Swine acute diarrhea syndrome coronavirus-induced apoptosis is caspase- and cyclophilin D-dependent. Emerg. Microbes Infect. 2020, 9, 439–456. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Collins, A.R. Induction of apoptosis in MRC-5, diploid human fetal lung cells after infection with human coronavirus OC43. Adv. Exp. Med. Biol. 2001, 494, 677–682. [Google Scholar] [PubMed] [Green Version]
- Kim, Y.; Lee, C. Porcine epidemic diarrhea virus induces caspase-independent apoptosis through activation of mitochondrial apoptosis-inducing factor. Virology 2014, 460–461, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Lee, L. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Razvi, E.S.; Welsh, R.M. Apoptosis in viral infections. Adv. Virus Res. 1995, 45, 1–60. [Google Scholar]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kim, J.; Park, S.; Kim, D.; Kim, M.; Baek, K.; Bae, J.Y.; Park, M.S.; Kim, W.K.; Lee, Y.; et al. MERS-CoV and SARS-CoV-2 replication can be inhibited by targeting the interaction between the viral spike protein and the nucleocapsid protein. Theranostics 2021, 11, 3853–3867. [Google Scholar] [CrossRef]
- Kim, D.; Maharjan, S.; Kim, J.; Park, S.; Park, J.A.; Park, B.K.; Lee, Y.; Kwon, H.J. MUC1-C influences cell survival in lung adenocarcinoma Calu-3 cells after SARS-CoV-2 infection. BMB Rep. 2021, 54, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Hammitt, L.L.; Kazungu, S.; Welch, S.; Bett, A.; Onyango, C.O.; Gunson, R.N.; Scott, J.A.; Nokes, D.J. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J. Clin. Microbiol. 2011, 49, 2318–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Favreau, D.J.; Meessen-Pinard, M.; Desforges, M.; Talbot, P.J. Human coronavirus-induced neuronal programmed cell death is cyclophilin d dependent and potentially caspase dispensable. J. Virol. 2012, 86, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meessen-Pinard, M.; Le Coupanec, A.; Desforges, M.; Talbot, P.J. Pivotal Role of Receptor-Interacting Protein Kinase 1 and Mixed Lineage Kinase Domain-Like in Neuronal Cell Death Induced by the Human Neuroinvasive Coronavirus OC43. J. Virol. 2016, 91, e01513-16. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Chen, Y.; Qi, S.; Shi, D.; Feng, L.; Sun, D. A Mini-Review on Cell Cycle Regulation of Coronavirus Infection. Front. Vet. Sci. 2020, 7, 586826. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [Green Version]
- Connolly, P.F.; Fearnhead, H.O. Viral hijacking of host caspases: An emerging category of pathogen-host interactions. Cell Death Differ. 2017, 24, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Koh, D.W.; Dawson, T.M.; Dawson, V.L. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol. Res. 2005, 52, 5–14. [Google Scholar] [CrossRef]
- Yang, K.; Hitomi, M.; Stacey, D.W. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006, 1, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Ma, J.; Su, M.; Shao, D.; Zhao, J.; Zhao, T.; Song, Z.; Meng, Y.; Jiao, P. Down-regulation of STAT3 induces the apoptosis and G1 cell cycle arrest in esophageal carcinoma ECA109 cells. Cancer Cell Int. 2018, 18, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Jang, Y.S.; Lee, H.J.; Lee, C.Y.; Shin, D.Y.; Oh, S.H. Inhibition of STAT3 signaling induces apoptosis and suppresses growth of lung cancer: Good and bad. Lab. Anim. Res. 2019, 35, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halupa, A.; Bailey, M.L.; Huang, K.; Iscove, N.N.; Levy, D.E.; Barber, D.L. A novel role for STAT1 in regulating murine erythropoiesis: Deletion of STAT1 results in overall reduction of erythroid progenitors and alters their distribution. Blood 2005, 105, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Choe, W.H.; Hiasa, Y.; Kamegaya, Y.; Blackard, J.T.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 2005, 128, 1034–1041. [Google Scholar] [CrossRef]
- Mizutani, T.; Fukushi, S.; Murakami, M.; Hirano, T.; Saijo, M.; Kurane, I.; Morikawa, S. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett. 2004, 577, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.; Wang, Y.; Zhou, X.; Long, J.E. STAT3 roles in viral infection: Antiviral or proviral? Future Virol. 2018, 13, 557–574. [Google Scholar] [CrossRef]
- Kaye, M. SARS-associated coronavirus replication in cell lines. Emerg. Infect. Dis. 2006, 12, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, D.W.; Thornberry, N.A. Caspases: Killer proteases. Trends Biochem Sci. 1997, 22, 299–306. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Ströher, U.; DiCaro, A.; Li, Y.; Strong, J.E.; Aoki, F.; Plummer, F.; Jones, S.M.; Feldmann, H. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-alpha. J. Infect. Dis. 2004, 189, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, Z.; Sun, J.; Zhang, Y.; Wei, C.; Ke, X.; Liu, Y.; Deng, L.; Wang, H. MiR-16-5p mediates a positive feedback loop in EV71-induced apoptosis and suppresses virus replication. Sci. Rep. 2017, 7, 16422. [Google Scholar] [CrossRef] [Green Version]
- Thomson, B.J. Viruses and apoptosis. Int. J. Exp. Pathol. 2001, 82, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Heylbroeck, C.; Balachandran, S.; Servant, M.J.; DeLuca, C.; Barber, G.N.; Lin, R.; Hiscott, J. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 2000, 74, 3781–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeganeh, B.; Ghavami, S.; Rahim, M.N.; Klonisch, T.; Halayko, A.J.; Coombs, K.M. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Gioti, K.; Kottaridi, C.; Voyiatzaki, C.; Chaniotis, D.; Rampias, T.; Beloukas, A. Animal Coronaviruses Induced Apoptosis. Life 2021, 11, 185. [Google Scholar] [CrossRef]
- Lai, Y.; Wang, M.; Cheng, A.; Mao, S.; Ou, X.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; et al. Regulation of Apoptosis by Enteroviruses. Front. Microbiol. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007, 365, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressy, C.; Droby, G.N.; Maldonado, B.D.; Steuerwald, N.; Grdzelishvili, V.Z. Cell Cycle Arrest in G2/M Phase Enhances Replication of Interferon-Sensitive Cytoplasmic RNA Viruses via Inhibition of Antiviral Gene Expression. J. Virol. 2019, 93, e01885-18. [Google Scholar] [CrossRef] [Green Version]
- De Martino, L.; Marfé, G.; Longo, M.; Fiorito, F.; Montagnaro, S.; Iovane, V.; Decaro, N.; Pagnini, U. Bid cleavage, cytochrome c release and caspase activation in canine coronavirus-induced apoptosis. Vet. Microbiol. 2010, 141, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, H.Y.; Liu, D.X. Induction of caspase-dependent apoptosis in cultured cells by the avian coronavirus infectious bronchitis virus. J. Virol. 2001, 75, 6402–6409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.P.; Li, H.S.; Cheung, M.C.; Chan, R.W.; Yuen, K.M.; Mok, C.K.; Nicholls, J.M.; Peiris, J.S.; Chan, M.C. Highly pathogenic avian influenza H5N1 virus delays apoptotic responses via activation of STAT3. Sci. Rep. 2016, 6, 28593. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Kar-Roy, A.; Kumari, S.; Mayor, S.; Jameel, S. The hepatitis E virus ORF3 protein modulates epidermal growth factor receptor trafficking, STAT3 translocation, and the acute-phase response. J. Virol. 2008, 82, 7100–7110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.K.; Kim, D.; Park, S.; Maharjan, S.; Kim, J.; Choi, J.K.; Akauliya, M.; Lee, Y.; Kwon, H.J. Differential Signaling and Virus Production in Calu-3 Cells and Vero Cells upon SARS-CoV-2 Infection. Biomol. Ther. 2021, 29, 273–281. [Google Scholar] [CrossRef]
- Bitzer, M.; Prinz, F.; Bauer, M.; Spiegel, M.; Neubert, W.J.; Gregor, M.; Schulze-Osthoff, K.; Lauer, U. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32). J. Virol. 1999, 73, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, S.; Kang, M.; Kim, J.; Kim, D.; Park, S.; Kim, M.; Baek, K.; Lee, Y.; Kwon, H.-J. Apoptosis Enhances the Replication of Human Coronavirus OC43. Viruses 2021, 13, 2199. https://doi.org/10.3390/v13112199
Maharjan S, Kang M, Kim J, Kim D, Park S, Kim M, Baek K, Lee Y, Kwon H-J. Apoptosis Enhances the Replication of Human Coronavirus OC43. Viruses. 2021; 13(11):2199. https://doi.org/10.3390/v13112199
Chicago/Turabian StyleMaharjan, Sony, Mijeong Kang, Jinsoo Kim, Dongbum Kim, Sangkyu Park, Minyoung Kim, Kyeongbin Baek, Younghee Lee, and Hyung-Joo Kwon. 2021. "Apoptosis Enhances the Replication of Human Coronavirus OC43" Viruses 13, no. 11: 2199. https://doi.org/10.3390/v13112199
APA StyleMaharjan, S., Kang, M., Kim, J., Kim, D., Park, S., Kim, M., Baek, K., Lee, Y., & Kwon, H. -J. (2021). Apoptosis Enhances the Replication of Human Coronavirus OC43. Viruses, 13(11), 2199. https://doi.org/10.3390/v13112199