
Peritoneal Administration of a Subunit Vaccine Encapsulated in a Nanodelivery System Not Only Augments Systemic Responses against SARS-CoV-2 but Also Stimulates Responses in the Respiratory Tract

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Animals
2.3. Production of Recombinant SARS-CoV-2 Spike Protein
2.4. Formulation and Characterization of Spike Glycoprotein-Loaded-N,N,N-Trimethyl Chitosan Nanoparticles (S-TMC NPs)
2.5. Uptake of S-TMC NPs by Phagocytic Cells
2.6. Animal Immunization and Specimen Collection
2.7. Antigen-Specific Antibodies Detected Using the ELISA Assay
2.8. The Virion-IgG ELISA
2.9. Antibody Neutralization Assay
2.10. Assessment of Vaccine-Induced Cellular Immune Responses
2.11. Statistical Analyses
3. Results
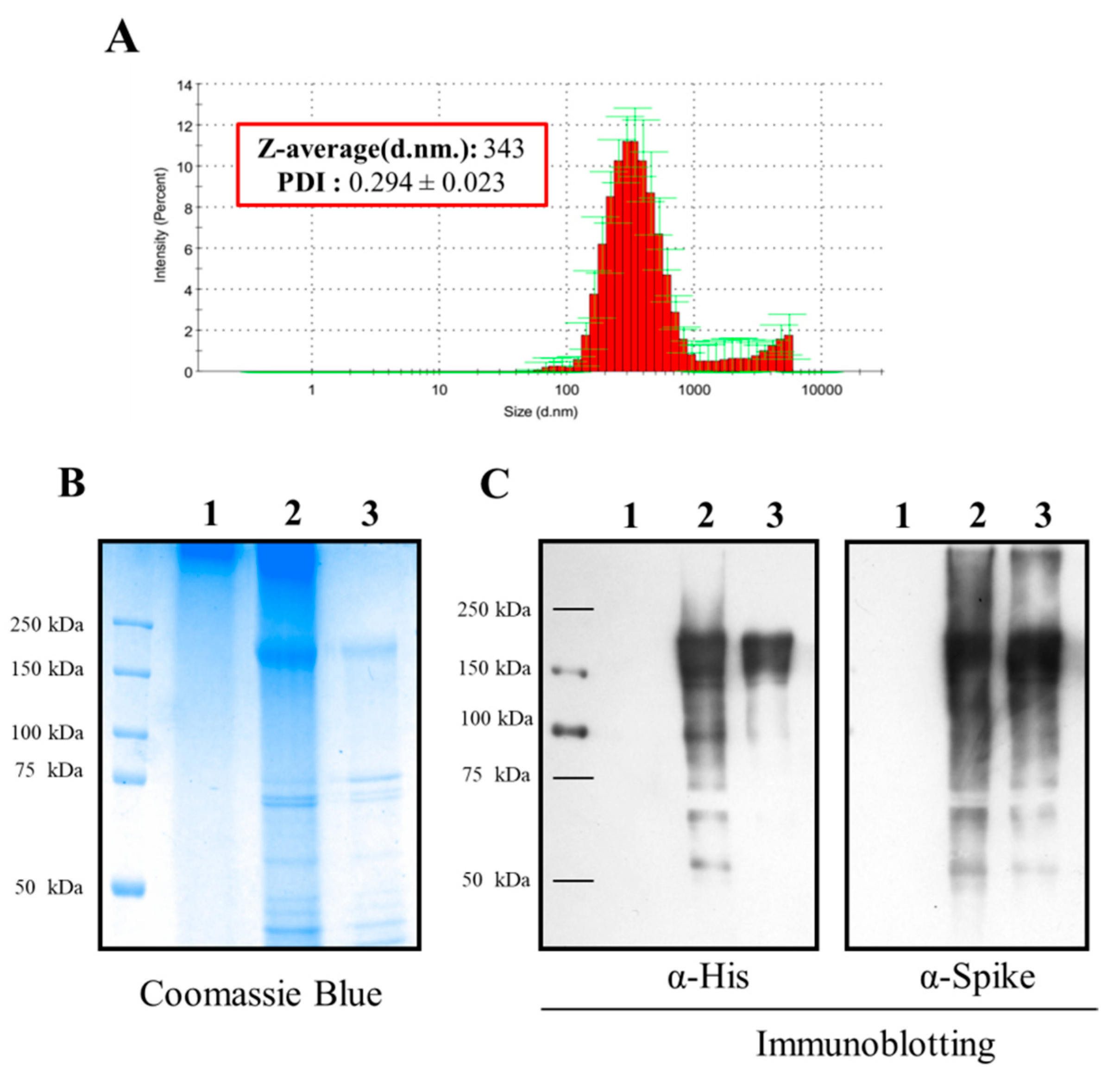
3.1. Formulation and In Vitro Characterization of S-TMC NPs
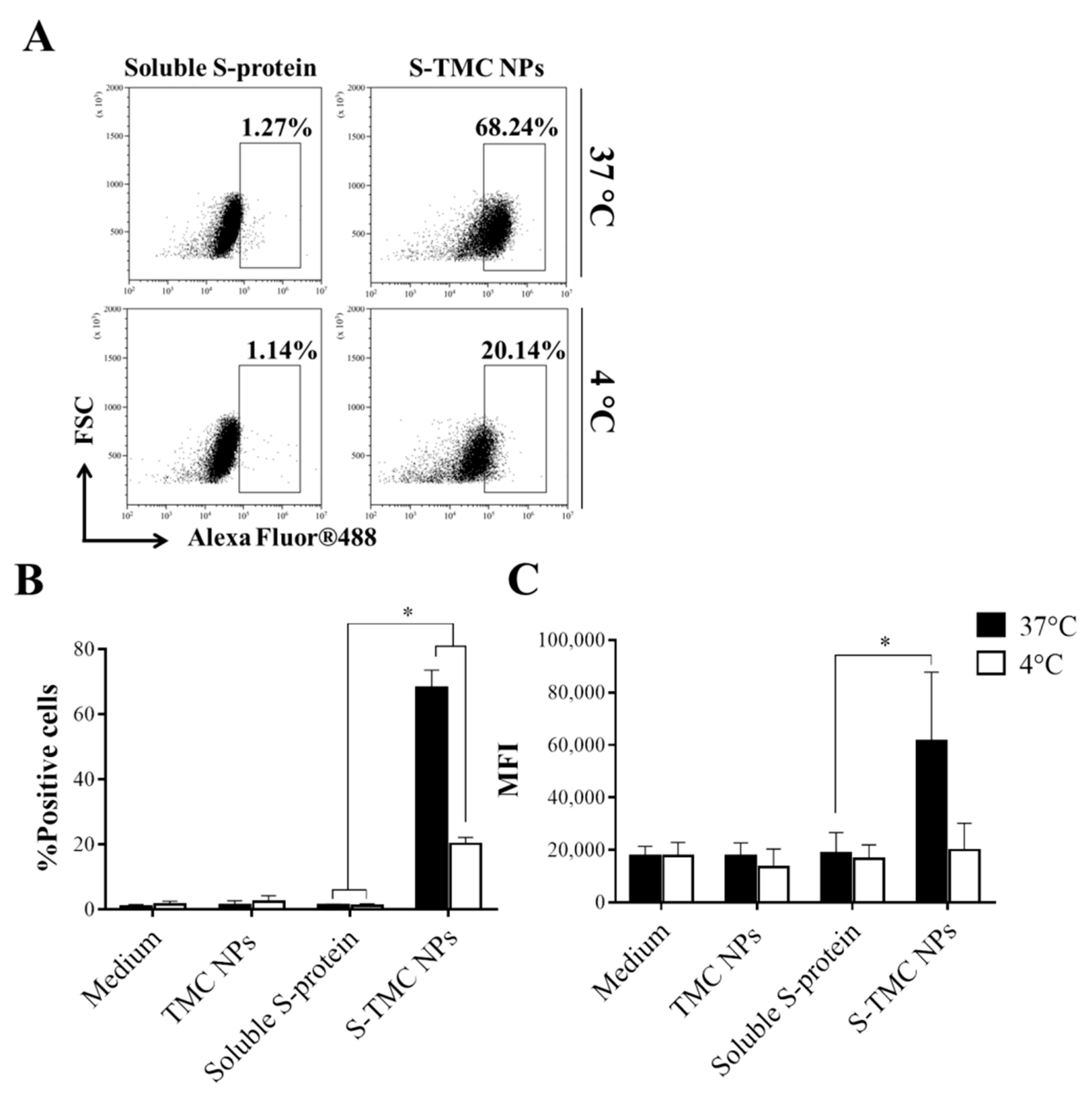
3.2. Uptake of S-TMC NPs by THP-1 Cells
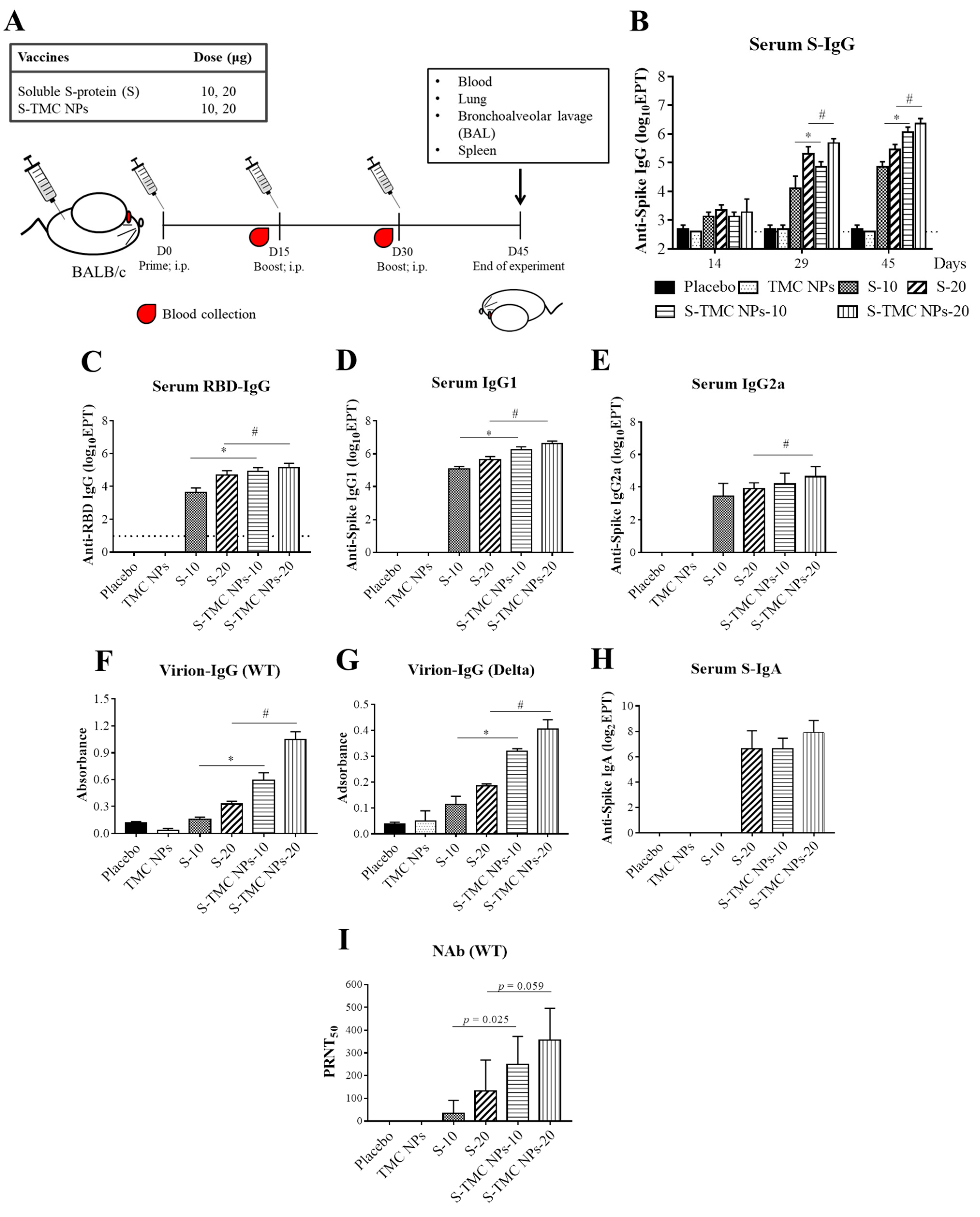
3.3. S-TMC NPs Strongly Induced the Circulating SARS-CoV-2 Antibody Response
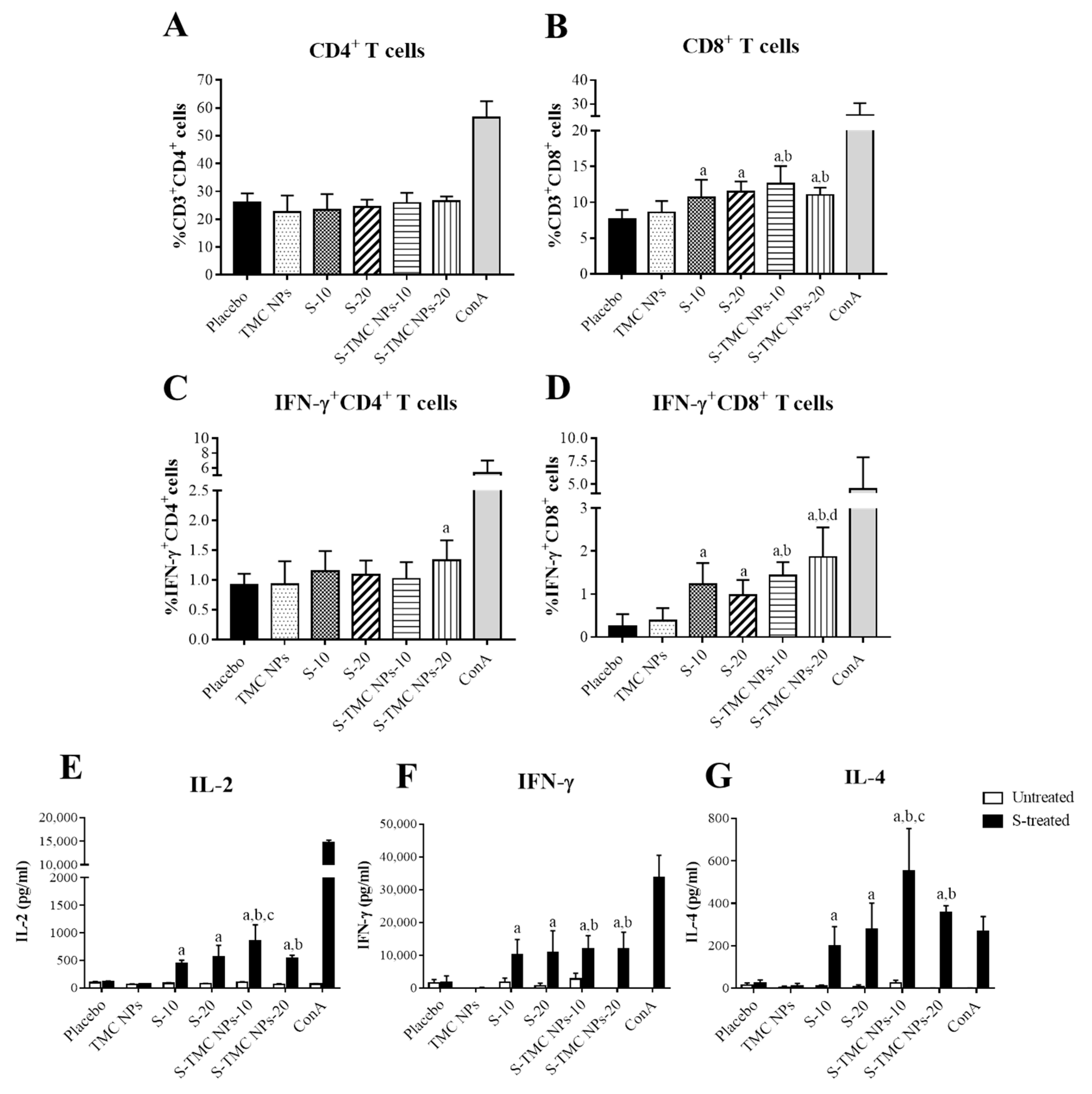
3.4. Immunization with S-Protein Nanoparticles Induced Systemic Cell-Mediated Immunity
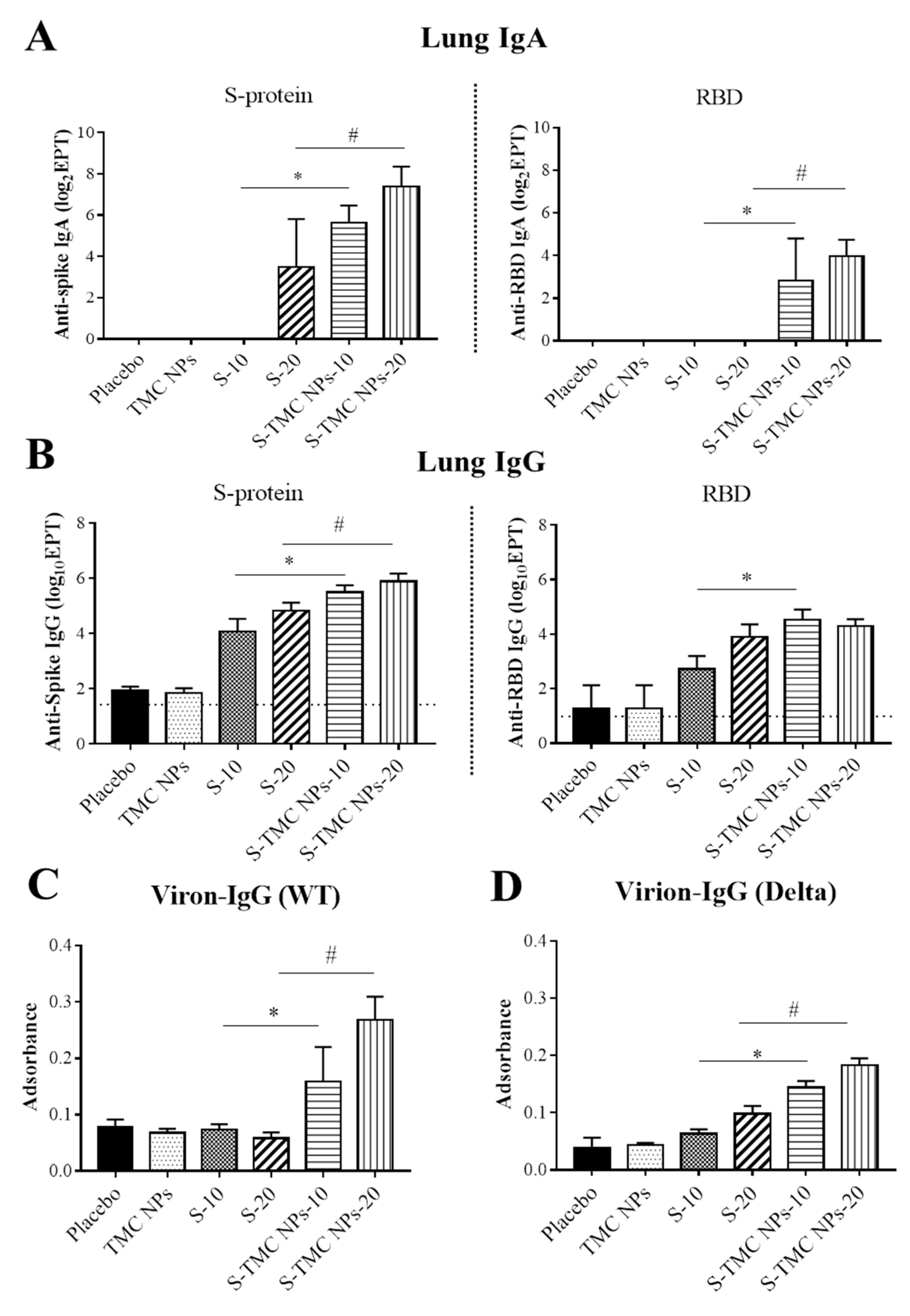
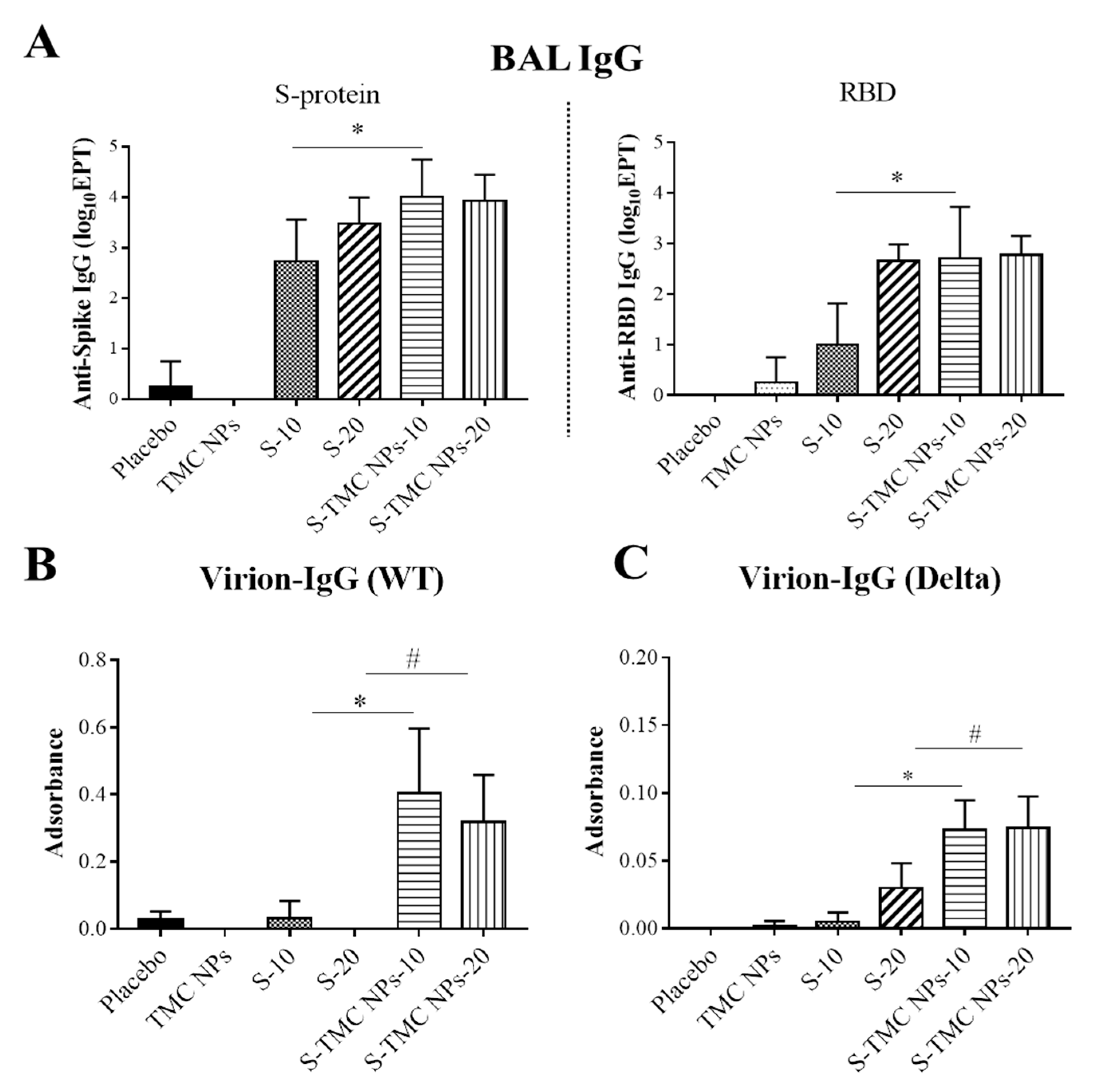
3.5. Intraperitoneal Administration of S-TMC NPs Stimulated Mucosal Immunity in the Respiratory Tract
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabibzadeh, A.; Esghaei, M.; Soltani, S.; Yousefi, P.; Taherizadeh, M.; Tameshkel, F.S.; Golahdooz, M.; Panahi, M.; Ajdarkosh, H.; Zamani, F.; et al. Evolutionary study of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as an emerging coronavirus: Phylogenetic analysis and literature review. Vet. Med. Sci. 2021, 7, 559–571. [Google Scholar] [CrossRef]
- Rastogi, M.; Pandey, N.; Shukla, A.; Singh, S.K. SARS coronavirus 2: From genome to infectome. Respir. Res. 2020, 21, 318. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- de Assis, R.R.; Jain, A.; Nakajima, R.; Jasinskas, A.; Felgner, J.; Obiero, J.M.; Norris, P.J.; Stone, M.; Simmons, G.; Bagri, A.; et al. Analysis of SARS-CoV-2 antibodies in COVID-19 convalescent blood using a coronavirus antigen microarray. Nat. Commun. 2021, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhang, L.; Chang, D.; Wang, J.; Hu, Y.; Chen, H.; Guo, L.; Wu, C.; Wang, C.; Wang, Y.; et al. The kinetics of humoral response and its relationship with the disease severity in COVID-19. Commun. Biol. 2020, 3, 780. [Google Scholar] [CrossRef]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.D.; et al. The origins and potential future of SARS-CoV-2 variants of concern in the evolving COVID-19 pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. Anti-Coronavirus Vaccines: Past Investigations on SARS-CoV-1 and MERS-CoV, the Approved Vaccines from BioNTech/Pfizer, Moderna, Oxford/AstraZeneca and others under Development Against SARS-CoV-2 Infection. Curr. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Haidere, M.F.; Ratan, Z.A.; Nowroz, S.; Zaman, S.B.; Jung, Y.J.; Hosseinzadeh, H.; Cho, J.Y. COVID-19 Vaccine: Critical Questions with Complicated Answers. Biomol. Ther. 2021, 29, 1–10. [Google Scholar] [CrossRef]
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 COVID-19 Vaccine. N. Engl. J. Med. 2021, 385, 1172–1183. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Li, X.; Min, M.; Du, N.; Gu, Y.; Hode, T.; Naylor, M.; Chen, D.; Nordquist, R.E.; Chen, W.R. Chitin, chitosan, and glycated chitosan regulate immune responses: The novel adjuvants for cancer vaccine. Clin. Dev. Immunol. 2013, 2013, 387023. [Google Scholar] [CrossRef]
- Mansouri, S.; Cuie, Y.; Winnik, F.; Shi, Q.; Lavigne, P.; Benderdour, M.; Beaumont, E.; Fernandes, J.C. Characterization of folate-chitosan-DNA nanoparticles for gene therapy. Biomaterials 2006, 27, 2060–2065. [Google Scholar] [CrossRef]
- Jearanaiwitayakul, T.; Sunintaboon, P.; Chawengkittikul, R.; Limthongkul, J.; Midoeng, P.; Warit, S.; Ubol, S. Nanodelivery system enhances the immunogenicity of dengue-2 nonstructural protein 1, DENV-2 NS1. Vaccine 2020, 38, 6814–6825. [Google Scholar] [CrossRef]
- Malik, A.; Gupta, M.; Mani, R.; Gogoi, H.; Bhatnagar, R. Trimethyl Chitosan Nanoparticles Encapsulated Protective Antigen Protects the Mice against Anthrax. Front. Immunol. 2018, 9, 562. [Google Scholar] [CrossRef]
- Tsai, M.H.; Chuang, C.C.; Chen, C.C.; Yen, H.J.; Cheng, K.M.; Chen, X.A.; Shyu, H.F.; Lee, C.Y.; Young, J.J.; Kau, J.H. Nanoparticles assembled from fucoidan and trimethylchitosan as anthrax vaccine adjuvant: In vitro and in vivo efficacy in comparison to CpG. Carbohydr. Polym. 2020, 236, 116041. [Google Scholar] [CrossRef]
- Dhakal, S.; Renu, S.; Ghimire, S.; Lakshmanappa, Y.S.; Hogshead, B.T.; Feliciano-Ruiz, N.; Lu, F.; HogenEsch, H.; Krakowka, S.; Lee, C.W.; et al. Mucosal Immunity and Protective Efficacy of Intranasal Inactivated Influenza Vaccine Is Improved by Chitosan Nanoparticle Delivery in Pigs. Front. Immunol. 2018, 9, 934. [Google Scholar] [CrossRef]
- Renu, S.; Feliciano-Ruiz, N.; Patil, V.; Schrock, J.; Han, Y.; Ramesh, A.; Dhakal, S.; Hanson, J.; Krakowka, S.; Renukaradhya, G.J. Immunity and Protective Efficacy of Mannose Conjugated Chitosan-Based Influenza Nanovaccine in Maternal Antibody Positive Pigs. Front. Immunol. 2021, 12, 584299. [Google Scholar] [CrossRef]
- Dabaghian, M.; Latifi, A.M.; Tebianian, M.; NajmiNejad, H.; Ebrahimi, S.M. Nasal vaccination with r4M2e.HSP70c antigen encapsulated into N-trimethyl chitosan (TMC) nanoparticulate systems: Preparation and immunogenicity in a mouse model. Vaccine 2018, 36, 2886–2895. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Arbeitman, C.R.; Auge, G.; Blaustein, M.; Bredeston, L.; Corapi, E.S.; Craig, P.O.; Cossio, L.A.; Dain, L.; D’Alessio, C.; Elias, F.; et al. Structural and functional comparison of SARS-CoV-2-spike receptor binding domain produced in Pichia pastoris and mammalian cells. Sci. Rep. 2020, 10, 21779. [Google Scholar] [CrossRef]
- Jearanaiwitayakul, T.; Seesen, M.; Chawengkirttikul, R.; Limthongkul, J.; Apichirapokey, S.; Sapsutthipas, S.; Phumiamorn, S.; Sunintaboon, P.; Ubol, S. Intranasal Administration of RBD Nanoparticles Confers Induction of Mucosal and Systemic Immunity against SARS-CoV-2. Vaccines 2021, 9, 768. [Google Scholar] [CrossRef]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef]
- Jeon, S.; Clavadetscher, J.; Lee, D.K.; Chankeshwara, S.V.; Bradley, M.; Cho, W.S. Surface Charge-Dependent Cellular Uptake of Polystyrene Nanoparticles. Nanomaterials 2018, 8, 1028. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, A.; Chen, K.; Chorny, A. Immunoglobulin responses at the mucosal interface. Annu. Rev. Immunol. 2011, 29, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zeng, W.; He, H.; Zhao, D.; Jiang, D.; Zhou, P.; Cheng, L.; Li, Y.; Ma, X.; Jin, T. Serum IgA, IgM, and IgG responses in COVID-19. Cell. Mol. Immunol. 2020, 17, 773–775. [Google Scholar] [CrossRef]
- Yu, H.Q.; Sun, B.Q.; Fang, Z.F.; Zhao, J.C.; Liu, X.Y.; Li, Y.M.; Sun, X.Z.; Liang, H.F.; Zhong, B.; Huang, Z.F.; et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur. Respir. J. 2020, 56, 2001526. [Google Scholar] [CrossRef]
- Russell, M.W.; Moldoveanu, Z.; Ogra, P.L.; Mestecky, J. Mucosal Immunity in COVID-19: A Neglected but Critical Aspect of SARS-CoV-2 Infection. Front. Immunol. 2020, 11, 611337. [Google Scholar] [CrossRef]
- Hassan, A.O.; Kafai, N.M.; Dmitriev, I.P.; Fox, J.M.; Smith, B.K.; Harvey, I.B.; Chen, R.E.; Winkler, E.S.; Wessel, A.W.; Case, J.B.; et al. A Single-Dose Intranasal ChAd Vaccine Protects Upper and Lower Respiratory Tracts against SARS-CoV-2. Cell 2020, 183, 169–184.e13. [Google Scholar] [CrossRef]
- Barker, K.A.; Etesami, N.S.; Shenoy, A.T.; Arafa, E.I.; de Ana, C.L.; Smith, N.M.; Martin, I.M.; Goltry, W.N.; Barron, A.M.; Browning, J.L.; et al. Lung-resident memory B cells protect against bacterial pneumonia. J. Clin. Investig. 2021, 131, e141810. [Google Scholar] [CrossRef]
- Kim, K.J.; Malik, A.B. Protein transport across the lung epithelial barrier. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L247–L259. [Google Scholar] [CrossRef] [Green Version]
- Spiekermann, G.M.; Finn, P.W.; Ward, E.S.; Dumont, J.; Dickinson, B.L.; Blumberg, R.S.; Lencer, W.I. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J. Exp. Med. 2002, 196, 303–310. [Google Scholar] [CrossRef]
- Offeddu, G.S.; Haase, K.; Gillrie, M.R.; Li, R.; Morozova, O.; Hickman, D.; Knutson, C.G.; Kamm, R.D. An on-chip model of protein paracellular and transcellular permeability in the microcirculation. Biomaterials 2019, 212, 115–125. [Google Scholar] [CrossRef]
- Abkar, M.; Fasihi-Ramandi, M.; Kooshki, H.; Lotfi, A.S. Oral immunization of mice with Omp31-loaded N-trimethyl chitosan nanoparticles induces high protection against Brucella melitensis infection. Int. J. Nanomed. 2017, 12, 8769–8778. [Google Scholar] [CrossRef] [Green Version]
- Najminejad, H.; Kalantar, S.M.; Mokarram, A.R.; Dabaghian, M.; Abdollahpour-Alitappeh, M.; Ebrahimi, S.M.; Tebianian, M.; Ramandi, M.F.; Sheikhha, M.H. Bordetella pertussis antigens encapsulated into N-trimethyl chitosan nanoparticulate systems as a novel intranasal pertussis vaccine. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2605–2611. [Google Scholar] [CrossRef]
- Mosafer, J.; Sabbaghi, A.H.; Badiee, A.; Dehghan, S.; Tafaghodi, M. Preparation, characterization and in vivo evaluation of alginate-coated chitosan and trimethylchitosan nanoparticles loaded with PR8 influenza virus for nasal immunization. Asian J. Pharm. Sci. 2019, 14, 216–221. [Google Scholar] [CrossRef]
- Jearanaiwitayakul, T.; Sunintaboon, P.; Chawengkittikul, R.; Limthongkul, J.; Midoeng, P.; Chaisuwirat, P.; Warit, S.; Ubol, S. Whole inactivated dengue virus-loaded trimethyl chitosan nanoparticle-based vaccine: Immunogenic properties in ex vivo and in vivo models. Hum. Vaccines Immunother. 2021, 17, 2793–2807. [Google Scholar] [CrossRef]
- Malik, A.; Gupta, M.; Gupta, V.; Gogoi, H.; Bhatnagar, R. Novel application of trimethyl chitosan as an adjuvant in vaccine delivery. Int. J. Nanomed. 2018, 13, 7959–7970. [Google Scholar] [CrossRef] [Green Version]
- Addetia, A.; Crawford, K.H.D.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Bloom, J.D.; Greninger, A.L. Neutralizing Antibodies Correlate with Protection from SARS-CoV-2 in Humans during a Fishery Vessel Outbreak with a High Attack Rate. J. Clin. Microbiol. 2020, 58, e02107-20. [Google Scholar] [CrossRef]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, X.; Zhou, Y.; Sun, J.; Liu, X.; Zhang, J.; Mei, X.; Zhong, J.; Zhao, J.; Ran, P. COVID-19 Severity Correlates with Weaker T-Cell Immunity, Hypercytokinemia, and Lung Epithelium Injury. Am. J. Respir. Crit. Care Med. 2020, 202, 606–610. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjug. Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef]
- García-Montero, C.; Fraile-Martínez, O.; Bravo, C.; Torres-Carranza, D.; Sanchez-Trujillo, L.; Gómez-Lahoz, A.M.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A.; Bujan, J.; et al. An Updated Review of SARS-CoV-2 Vaccines and the Importance of Effective Vaccination Programs in Pandemic Times. Vaccines 2021, 9, 433. [Google Scholar] [CrossRef]
- Mohandas, S.; Yadav, P.D.; Shete-Aich, A.; Abraham, P.; Vadrevu, K.M.; Sapkal, G.; Mote, C.; Nyayanit, D.; Gupta, N.; Srinivas, V.K.; et al. Immunogenicity and protective efficacy of BBV152, whole virion inactivated SARS- CoV-2 vaccine candidates in the Syrian hamster model. Iscience 2021, 24, 102054. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.W.; Zhang, Y.; Zhou, D.H.; Chen, Y.Q.; Yang, J.Y.; Yan, H.M. PIKA provides an adjuvant effect to induce strong mucosal and systemic humoral immunity against SARS-CoV. Virol. Sin. 2011, 26, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.; Dixit, S.; Verma, R.; Duncan, S.A.; Smith, L.; Giambartolomei, G.H.; Singh, S.R.; Dennis, V.A. Encapsulation of Recombinant MOMP in Extended-Releasing PLGA 85:15 Nanoparticles Confer Protective Immunity Against a Chlamydia muridarum Genital Challenge and Re-Challenge. Front. Immunol. 2021, 12, 660932. [Google Scholar] [CrossRef]
- Stokes, C.R.; Soothill, J.F.; Turner, M.W. Immune exclusion is a function of IgA. Nature 1975, 255, 745–746. [Google Scholar] [CrossRef]
- Ejemel, M.; Li, Q.; Hou, S.; Schiller, Z.A.; Tree, J.A.; Wallace, A.; Amcheslavsky, A.; Kurt Yilmaz, N.; Buttigieg, K.R.; Elmore, M.J.; et al. A cross-reactive human IgA monoclonal antibody blocks SARS-CoV-2 spike-ACE2 interaction. Nat. Commun. 2020, 11, 4198. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Vibholm, L.K.; Monrad, I.; Olesen, R.; Frattari, G.S.; Pahus, M.H.; Højen, J.F.; Gunst, J.D.; Erikstrup, C.; Holleufer, A.; et al. SARS-CoV-2 elicits robust adaptive immune responses regardless of disease severity. EBioMedicine 2021, 68, 103410. [Google Scholar] [CrossRef]
- Chen, K.; Magri, G.; Grasset, E.K.; Cerutti, A. Rethinking mucosal antibody responses: IgM, IgG and IgD join IgA. Nat. Rev. Immunol. 2020, 20, 427–441. [Google Scholar] [CrossRef]
- Vissers, M.; Ahout, I.M.; de Jonge, M.I.; Ferwerda, G. Mucosal IgG Levels Correlate Better with Respiratory Syncytial Virus Load and Inflammation than Plasma IgG Levels. Clin. Vaccine Immunol. 2015, 23, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Robert-Guroff, M. IgG surfaces as an important component in mucosal protection. Nat. Med. 2000, 6, 129–130. [Google Scholar] [CrossRef]
- Bai, Y.; Ye, L.; Tesar, D.B.; Song, H.; Zhao, D.; Björkman, P.J.; Roopenian, D.C.; Zhu, X. Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proc. Natl. Acad. Sci. USA 2011, 108, 18406–18411. [Google Scholar] [CrossRef] [Green Version]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jearanaiwitayakul, T.; Apichirapokey, S.; Chawengkirttikul, R.; Limthongkul, J.; Seesen, M.; Jakaew, P.; Trisiriwanich, S.; Sapsutthipas, S.; Sunintaboon, P.; Ubol, S. Peritoneal Administration of a Subunit Vaccine Encapsulated in a Nanodelivery System Not Only Augments Systemic Responses against SARS-CoV-2 but Also Stimulates Responses in the Respiratory Tract. Viruses 2021, 13, 2202. https://doi.org/10.3390/v13112202
Jearanaiwitayakul T, Apichirapokey S, Chawengkirttikul R, Limthongkul J, Seesen M, Jakaew P, Trisiriwanich S, Sapsutthipas S, Sunintaboon P, Ubol S. Peritoneal Administration of a Subunit Vaccine Encapsulated in a Nanodelivery System Not Only Augments Systemic Responses against SARS-CoV-2 but Also Stimulates Responses in the Respiratory Tract. Viruses. 2021; 13(11):2202. https://doi.org/10.3390/v13112202
Chicago/Turabian StyleJearanaiwitayakul, Tuksin, Suttikarn Apichirapokey, Runglawan Chawengkirttikul, Jitra Limthongkul, Mathurin Seesen, Phissinee Jakaew, Sakalin Trisiriwanich, Sompong Sapsutthipas, Panya Sunintaboon, and Sukathida Ubol. 2021. "Peritoneal Administration of a Subunit Vaccine Encapsulated in a Nanodelivery System Not Only Augments Systemic Responses against SARS-CoV-2 but Also Stimulates Responses in the Respiratory Tract" Viruses 13, no. 11: 2202. https://doi.org/10.3390/v13112202
APA StyleJearanaiwitayakul, T., Apichirapokey, S., Chawengkirttikul, R., Limthongkul, J., Seesen, M., Jakaew, P., Trisiriwanich, S., Sapsutthipas, S., Sunintaboon, P., & Ubol, S. (2021). Peritoneal Administration of a Subunit Vaccine Encapsulated in a Nanodelivery System Not Only Augments Systemic Responses against SARS-CoV-2 but Also Stimulates Responses in the Respiratory Tract. Viruses, 13(11), 2202. https://doi.org/10.3390/v13112202