
Molecular Analysis of the Avian H7 Influenza Viruses Circulating in South Korea during 2018–2019: Evolutionary Significance and Associated Zoonotic Threats


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Virus Isolation
2.2. RNA Extraction and Next-Generation Sequencing (NGS)
2.3. Phylogenetic Analysis
2.4. Temporal Dynamics of the H7 Isolates
2.5. Accession Numbers
3. Results
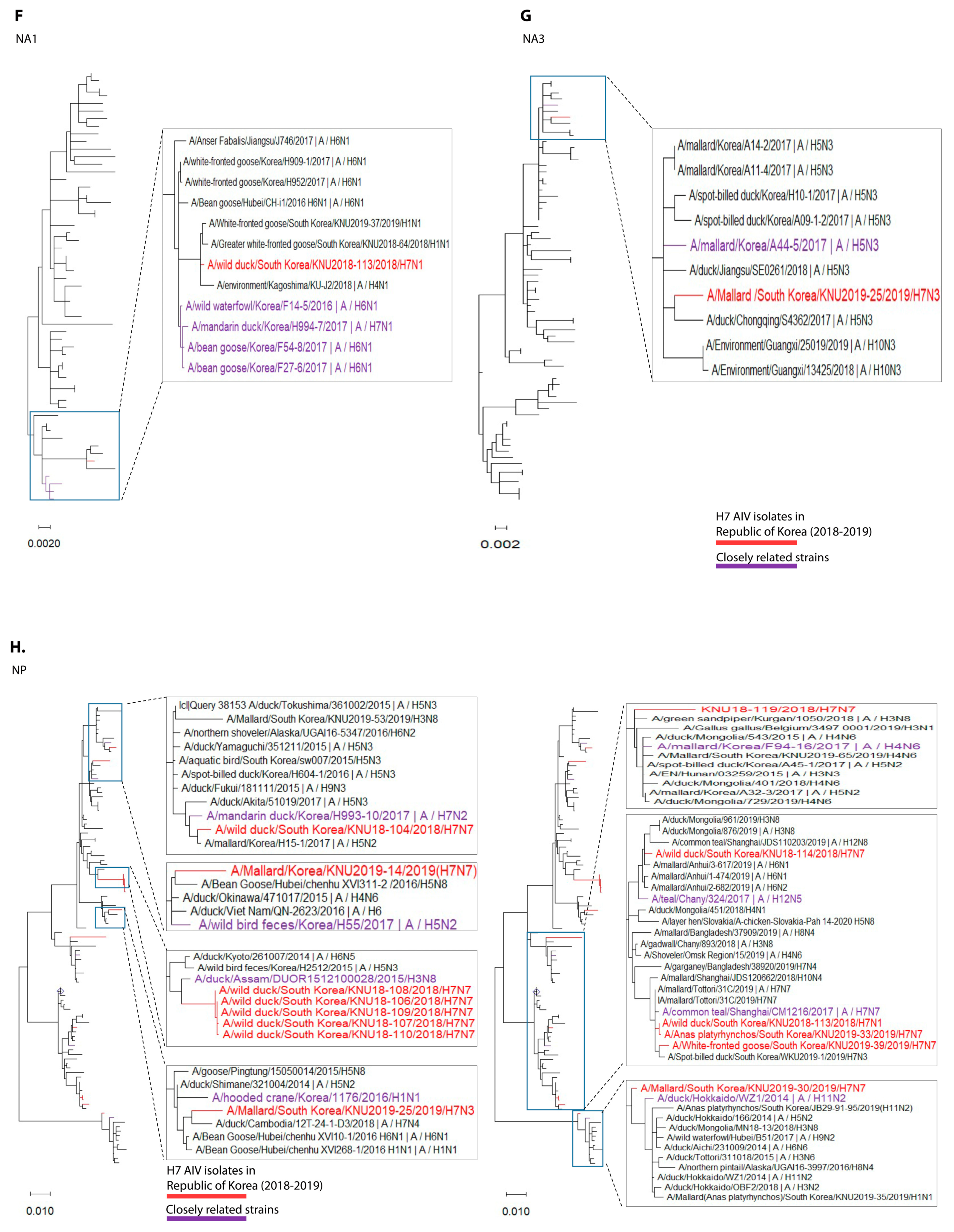
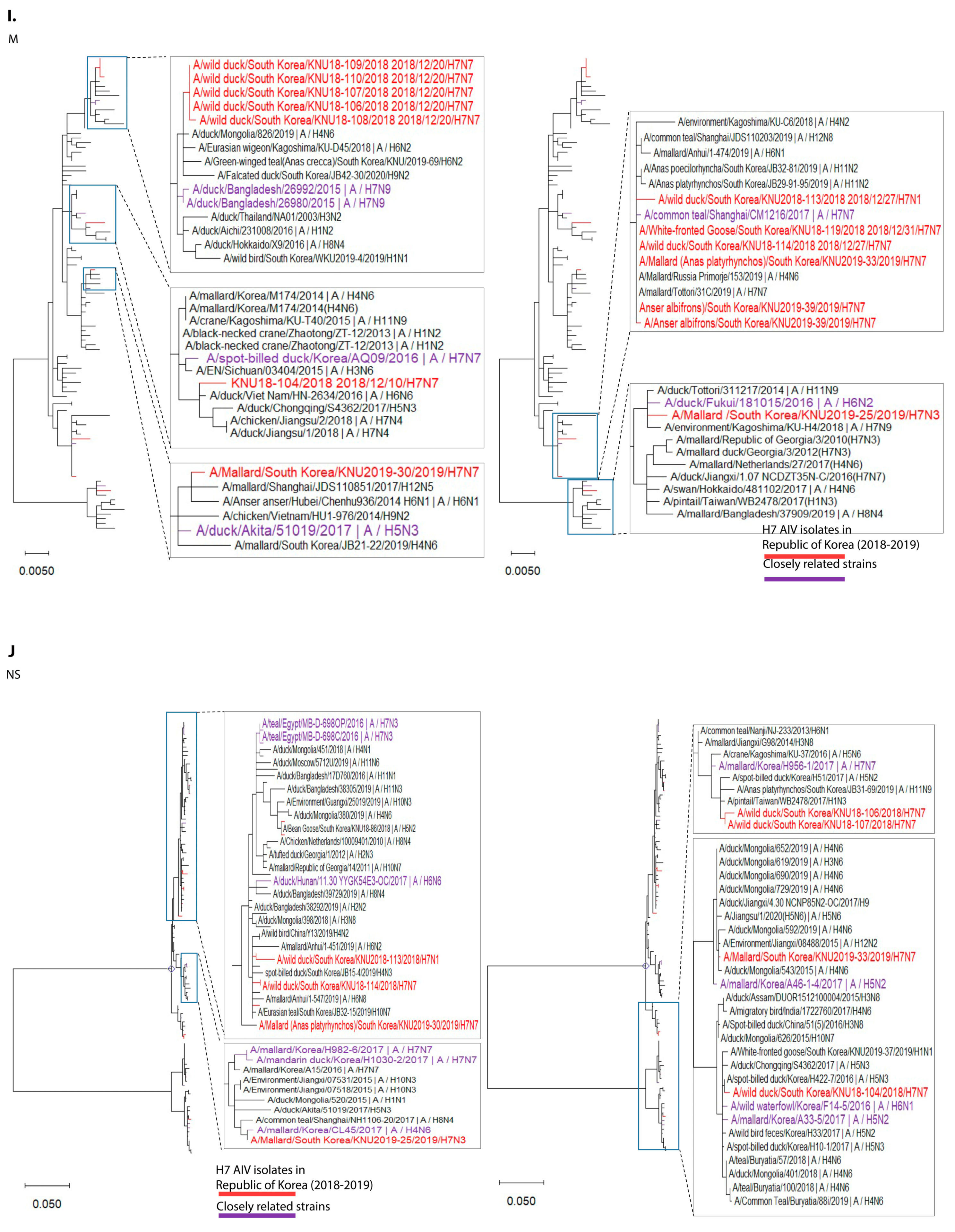
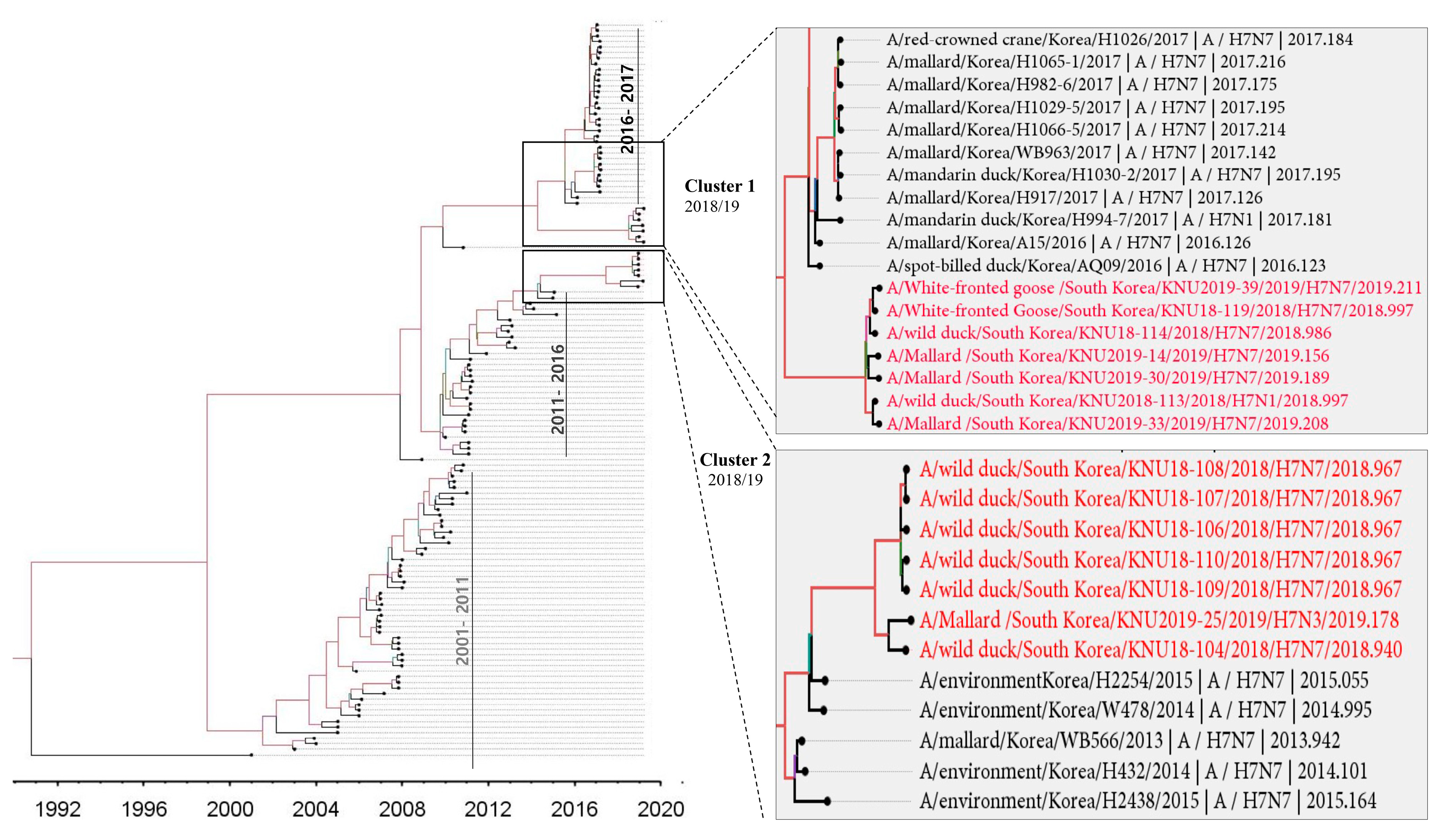
3.1. Isolation and Genomic Characterization of H7 IAVs during 2018–2019 in the Republic of Korea
3.2. Molecular Characteristics of the H7 Isolates of 2018–2019 from the Republic of Korea
3.3. Hypothesis for Reassortment Event in Each Gene Segment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Webby, R.J.; Webster, R.G. Emergence of influenza A viruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1817–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Capua, I.; Alexander, D.J. Avian influenza infections in birds—A moving target. Influenza Other Respir. Viruses 2007, 1, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfanti, L.; Monne, I.; Tamba, M.; Santucci, U.; Massi, P.; Patregnani, T.; Loli Piccolomini, L.; Natalini, S.; Ferri, G.; Cattoli, G.; et al. Highly pathogenic H7N7 avian influenza in Italy. Vet. Rec. 2014, 174, 382. [Google Scholar] [CrossRef]
- Van Gils, J.A.; Munster, V.J.; Radersma, R.; Liefhebber, D.; Fouchier, R.A.; Klaassen, M. Hampered foraging and migratory performance in swans infected with low-pathogenic avian influenza A virus. PLoS ONE 2007, 2, e184. [Google Scholar] [CrossRef]
- Joseph, U.; Su, Y.C.; Vijaykrishna, D.; Smith, G.J. The ecology and adaptive evolution of influenza A interspecies transmission. Influenza Other Respir. Viruses 2017, 11, 74–84. [Google Scholar] [CrossRef]
- Abdelwhab, E.M.; Veits, J.; Mettenleiter, T.C. Prevalence and control of H7 avian influenza viruses in birds and humans. Epidemiol. Infect. 2014, 142, 896–920. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Schneeberger, P.M.; Rozendaal, F.W.; Broekman, J.M.; Kemink, S.A.; Munster, V.; Kuiken, T.; Rimmelzwaan, G.F.; Schutten, M.; Van Doornum, G.J.; et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 1356–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, C.; Mok, C.K.P.; Zhu, W.; Zhou, H.; He, J.; Guan, W.; Wu, J.; Song, W.; Wang, D.; Liu, J.; et al. Human infection with highly pathogenic avian influenza A(H7N9) virus, China. Emerg. Infect. Dis. 2017, 23, 1332–1340. [Google Scholar] [CrossRef] [Green Version]
- Horimoto, T.; Kawaoka, Y. Pandemic threat posed by avian influenza A viruses. Clin. Microbiol. Rev. 2001, 14, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xiong, C.; Chen, J.; Chen, G.; Zhang, J.; Li, Y.; Xiong, Y.; Wang, R.; Cao, Y.; Chen, Q.; et al. Two genetically diverse H7N7 avian influenza viruses isolated from migratory birds in central China. Emerg. Microbes Infect. 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Tang, W.; Li, X.; Tang, L.; Wang, T.; He, G. Characterization of the low-pathogenic H7N7 avian influenza virus in Shanghai, China. Poult. Sci. 2021, 100, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, Y.J.; Park, C.K.; Oem, J.K.; Lee, O.S.; Kang, H.M.; Choi, J.G.; Bae, Y.C. Highly pathogenic avian influenza (H5N1) outbreaks in wild birds and poultry, South Korea. Emerg. Infect. Dis. 2012, 18, 480–483. [Google Scholar] [CrossRef]
- Lee, Y.N.; Cheon, S.H.; Lee, E.K.; Heo, G.B.; Bae, Y.C.; Joh, S.J.; Lee, M.H.; Lee, Y.J. Pathogenesis and genetic characteristics of novel reassortant low-pathogenic avian influenza H7 viruses isolated from migratory birds in the Republic of Korea in the winter of 2016–2017. Emerg. Microbes Infect. 2018, 7, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwood, P.F.; Karlsson, E.A.; Horm, S.V.; Ly, S.; Heng, S.; Chin, S.; Darapheak, C.; Saunders, D.; Chanthap, L.; Rith, S.; et al. Circulation and characterization of seasonal influenza viruses in Cambodia, 2012–2015. Influenza Respir. Viruses 2019, 13, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Trinh, T.-T.T.; Tiwari, I.; Durairaj, K.; Duong, B.T.; Nguyen, A.T.V.; Tuong, H.T.; Hoang, V.T.; Than, D.D.; Nam, S.; Yeo, S.-J.; et al. Genetic characterization and pathogenesis of avian influenza virus H7N3 isolated from spot-billed ducks in South Korea, Early 2019. Viruses 2021, 13, 856. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 2001, 293, 1840–1842. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, L.; Yan, Z.; Gan, T.; Li, L.; Chen, R.; Chen, R.; Zheng, Z.; Hong, W.; Wang, J.; et al. Dual E627K and D701N mutations in the PB2 protein of A(H7N9) influenza virus increased its virulence in mammalian models. Sci. Rep. 2015, 5, 14170. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef] [Green Version]
- Hulse-Post, D.J.; Franks, J.; Boyd, K.; Salomon, R.; Hoffmann, E.; Yen, H.L.; Webby, R.J.; Walker, D.; Nguyen, T.D.; Webster, R.G. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J. Virol. 2007, 81, 8515–8524. [Google Scholar] [CrossRef] [Green Version]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar] [CrossRef]
- Leung, B.W.; Chen, H.; Brownlee, G.G. Correlation between polymerase activity and pathogenicity in two duck H5N1 influenza viruses suggests that the polymerase contributes to pathogenicity. Virology 2010, 401, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Mei, K.; Liu, G.; Chen, Z.; Gao, Z.; Zhao, L.; Jin, T.; Yu, X.; Chen, Q. Deep sequencing reveals the viral adaptation process of environment-derived H10N8 in mice. Infect. Genet. Evol. 2016, 37, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, X.; Gao, W.; Wang, C.; Wang, J.; Sun, H.; Sun, Y.; Guo, L.; Zhang, R.; Chang, K.-C.; et al. Prevailing PA Mutation K356R in Avian Influenza H9N2 Virus Increases Mammalian Replication and Pathogenicity. J. Virol. 2016, 90, 8105–8114. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.; Cooper, L.; Xu, X.; Thompson, W.; Krauss, S.; Guan, Y.; Zhou, N.; Klimov, A.; Cox, N.; Webster, R.; et al. Molecular changes associated with the transmission of avian influenza a H5N1 and H9N2 viruses to humans. J. Med Virol. 2002, 66, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Nidom, C.A.; Takano, R.; Yamada, S.; Sakai-Tagawa, Y.; Daulay, S.; Aswadi, D.; Suzuki, T.; Suzuki, Y.; Shinya, K.; Iwatsuki-Horimoto, K.; et al. Influenza A (H5N1) viruses from pigs, Indonesia. Emerg. Infect. Dis. 2010, 16, 1515–1523. [Google Scholar] [CrossRef]
- Tabynov, K.; Sansyzbay, A.; Sandybayev, N.; Mambetaliyev, M. The pathogenicity of swan derived H5N1 virus in birds and mammals and its gene analysis. Virol. J. 2014, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- McKimm-Breschkin, J.L.; Sahasrabudhe, A.; Blick, T.J.; McDonald, M.; Colman, P.M.; Hart, G.J.; Bethell, R.C.; Varghese, J.N. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2en-derived inhibitors. J. Virol. 1998, 72, 2456–2462. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-Y.A.; Rijal, P.; Schimanski, L.; Powell, T.J.; Lin, T.-Y.; McCauley, J.W.; Daniels, R.S.; Townsend, A.R. Focused antibody response to influenza linked to antigenic drift. J. Clin. Investig. 2015, 125, 2631–2645. [Google Scholar] [CrossRef]
- Han, X.; Bertzbach, L.D.; Veit, M. Mimicking the passage of avian influenza viruses through the gastrointestinal tract of chickens. Vet. Microbiol. 2019, 239, 108462. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Capua, I.; Alexander, D.J. Avian influenza: Recent developments. Avian Pathol. 2004, 33, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Javakhishvili, Z.; Russell, C.A.; Machablishvili, A.; Lexmond, P.; Verhagen, J.H.; Vuong, O.; Onashvili, T.; Donduashvili, M.; Smith, D.J.; et al. Avian influenza virus surveillance in wild birds in Georgia: 2009–2011. PLoS ONE 2013, 8, e58534. [Google Scholar] [CrossRef] [Green Version]
- Naeve, C.W.; Webster, R.G. Sequence of the hemagglutinin gene from influenza virus A/Seal/Mass/1/80. Virology 1983, 129, 298–308. [Google Scholar] [CrossRef]
- Zhao, H.; Chu, H.; Zhao, X.; Shuai, H.; Wong, B.H.-Y.; Wen, L.; Yuan, S.; Zheng, B.-J.; Zhou, J.; Yuen, K.-Y. Novel residues in the PA protein of avian influenza H7N7 virus affect virulence in mammalian hosts. Virology 2016, 498, 4161–4164. [Google Scholar] [CrossRef]
- Saito, T.; Tanikawa, T.; Uchida, Y.; Takemae, N.; Kanehira, K.; Tsunekuni, R. Intracontinental and intercontinental dissemination of Asian H5 highly pathogenic avian influenza virus (clade 2.3.4.4) in the winter of 2014–2015. Rev. Med. Virol. 2015, 25, 388–405. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Sub-Type | PB2 | PB1 | PB1-F2 | PA | HA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L89V | E627K | D701N | L13P | R207K | N66S | H266R | F277S | K356R | S409N | Cleavage Site | S138A | Q226L | ||
| Markers of Mammalian Adaptation [17] | Increased Virulence in Mice [18] | Increased Polymerase Activity, Increased Virulence [19,20] | Increased virulence in Mammals [21] | Increased Polymerase Activity, Increased Virulence [22] | Increased Virulence, Mammalian Adaptation [23] | Increased Polymerase Activity, Increased Virulence in Mammals and Birds [24] | Enhanced Transmission, Marker of Mammalian Adaptation [25] | Increased Virus Binding to Human-Type Receptors [26] | ||||||
| A/Italy/3/2013 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| A/Jiangsu/1/2018 | H7N4 | V | K | D | P | K | N | R | S | K | S | PKRRERR↓GLF | A | Q |
| A/Shanghai/1/2013 | H7N9 | V | K | D | P | K | N | R | S | R | N | PELPKGR↓GLF | S | Q |
| KNU2018-104 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-106 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-107 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-108 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-109 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-110 | H7N7 | V | E | D | P | K | N | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-113 | H7N1 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-114 | H7N7 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2018-119 | H7N7 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2019-14 | H7N7 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2019-33 | H7N7 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2019-39 | H7N7 | V | E | D | P | K | S | R | S | K | S | PELPKGR↓GLF | A | Q |
| KNU2019-25 | H7N3 | V | E | D | P | K | N | R | F | K | S | PELPKGR↓GLF | A | Q |
| KNU2019-30 | H7N7 | V | E | D | P | K | S | R | S | K | S | PEPPKGR↓GLF | A | Q |
| Virus | Sub-Type | NP | NA | M1 | M2 | NS1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Virulence | ||||||||||||||
| V41I | A184K | D210E | M26I | I107V | R294K | V15I | N30D | T215A | I28V | S31N | L55F | P/A42S | ||
| Enhanced RNP Activity [18] | Increased Virulence [27] | Enhanced RNP Activity [18] | Increased Virulence in Mammals | Resistant to Oseltamivir & Zanamivir Resistent [28] | Increased Virulence in Mice [29] | Mammalian Adaptation [30] | Increased Virulence in Mice | |||||||
| A/Italy/3/2013 | H7N7 | I | K | E | I | I | R | V | D | A | I | S | L | S |
| A/Jiangsu/1/2018 | H7N4 | I | K | E | L | I | R | V | D | A | I | S | L | S |
| A/Shanghai/1/2013 | H7N9 | I | K | E | I | V | K | I | D | A | V | N | F | S |
| KNU2018-104 | H7N7 | I | K | E | I | V | R | V | D | A | V | S | L | A |
| KNU2018-106 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-107 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-108 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-109 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-110 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-113 | H7N1 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-114 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2018-119 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2019-14 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2019-33 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | A |
| KNU2019-39 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2019-25 | H7N3 | I | K | E | I | V | R | V | D | A | I | S | L | S |
| KNU2019-30 | H7N7 | I | K | E | I | V | R | V | D | A | I | S | L | S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, B.T.; Bal, J.; Sung, H.W.; Yeo, S.-J.; Park, H. Molecular Analysis of the Avian H7 Influenza Viruses Circulating in South Korea during 2018–2019: Evolutionary Significance and Associated Zoonotic Threats. Viruses 2021, 13, 2260. https://doi.org/10.3390/v13112260
Duong BT, Bal J, Sung HW, Yeo S-J, Park H. Molecular Analysis of the Avian H7 Influenza Viruses Circulating in South Korea during 2018–2019: Evolutionary Significance and Associated Zoonotic Threats. Viruses. 2021; 13(11):2260. https://doi.org/10.3390/v13112260
Chicago/Turabian StyleDuong, Bao Tuan, Jyotiranjan Bal, Haan Woo Sung, Seon-Ju Yeo, and Hyun Park. 2021. "Molecular Analysis of the Avian H7 Influenza Viruses Circulating in South Korea during 2018–2019: Evolutionary Significance and Associated Zoonotic Threats" Viruses 13, no. 11: 2260. https://doi.org/10.3390/v13112260
APA StyleDuong, B. T., Bal, J., Sung, H. W., Yeo, S. -J., & Park, H. (2021). Molecular Analysis of the Avian H7 Influenza Viruses Circulating in South Korea during 2018–2019: Evolutionary Significance and Associated Zoonotic Threats. Viruses, 13(11), 2260. https://doi.org/10.3390/v13112260