
Full Viral Genome Sequencing and Phylogenomic Analysis of Feline Herpesvirus Type 1 (FHV-1) in Cheetahs (Acinonyx jubatus)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Host Animals
2.2. Viral Isolation
2.3. Viral DNA Extraction
2.4. Sequencing
2.5. Genome Assembly
2.6. Viral Genome Alignment
2.7. Variant Analysis
2.8. Phylogenetic and Recombination Analysis
2.9. Sequence Accession Numbers
3. Results
3.1. Sequencing and Genome Assembly
3.2. Variant Analysis
3.3. Phylogenetic and Recombination Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durant, S.; Mitchell, N.; Ipavec, A.; Groom, R. Acinonyx jubatus. The IUCN Red List of Threatened Species 2015: e. T219A50649567. Available online: https://www.iucnredlist.org/species/219/50649567 (accessed on 15 November 2021).
- Evermann, J.F.; Laurenson, M.K.; McKeirnan, A.J.; Caro, T.M. Infectious disease surveillance in captive and free-living cheetahs: An integral part of the species survival plan. Zoo Biol. 1993, 12, 125–133. [Google Scholar] [CrossRef]
- Witte, C.L.; Lamberski, N.; Rideout, B.A.; Vaida, F.; Citino, S.B.; Barrie, M.T.; Haefele, H.J.; Junge, R.E.; Murray, S.; Hungerford, L.L. Epidemiology of clinical feline herpesvirus infection in zoo-housed cheetahs (Acinonyx jubatus). J. Am. Vet. Med Assoc. 2017, 251, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Gaskell, R.; Dawson, S.; Radford, A.; Thiry, E. Feline herpesvirus. Vet. Res. 2007, 38, 337–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, L.; Marker, L.; Dubovi, E.; Spencer, J.A.; Evermann, J.F.; O’Brien, S.J. Serosurvey of viral infections in free-ranging Namibian cheetahs (Acinonyx jubatus). J. Wildl. Dis. 2004, 40, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, L.; Wack, R.; Duncan, M.; Montali, R.J.; Boon, D.; Stalis, I.; Crawshaw, G.J.; Cameron, K.N.; Mortenson, J.; Citino, S.; et al. Chronic eosinophilic dermatitis associated with persistent feline herpes virus infection in cheetahs (Acinonyx jubatus). Vet. Pathol. 2004, 41, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Gaskell, R.; Willoughby, K. Herpesviruses of carnivores. Vet. Microbiol. 1999, 69, 73–88. [Google Scholar] [CrossRef]
- Spencer, J.A.; Burroughs, R. Antibody response of captive cheetahs to modified-live feline virus vaccine. J. Wildl. Dis. 1991, 27, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, C.; Lamberski, N.; Rideout, B.; Fields, V.; Teare, C.; Barrie, M.; Hungerford, L. Development of a Case Definition for Clinical Feline Herpesvirus Infection in Cheetahs (Acinonyx jubatus) Housed in Zoos. J. Zoo Wildl. Med. 2013, 44, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Pennings, A.; Seeley, K.; Mathieu, A.; Foust, A.; Garner, M.M.; Ramer, J. Feline Herpesvirus Infection in Four Captive Cheetahs (Acinonyx jubatus) Postvaccinaiton. J. Zoo Wildl. Med. 2020, 51, 210–216. [Google Scholar] [CrossRef]
- Lamberski, N. Updated Vaccination Recommendations for Carnivores. In Fowler’s Zoo and Wild Animal Medicine; Elsevier: St. Louis, MO, USA, 2012; pp. 442–450. [Google Scholar]
- Woc Colburn, A.M.; Sanchez, C.R.; Citino, S.; Crosier, A.E.; Murray, S.; Kaandorp, J.; Kaandorp, C.; Marker, L. Clinical Management of Captive Cheetahs. In Cheetahs: Biology and Conservation; Academic Press: Cambridge, MA, USA, 2018; Volume 1, p. 12. [Google Scholar]
- Torres, R.S.G.; Hernández, B.E.A.; Helmick, K. Post-Vaccination Viral Disease in Two Cheetah (Acinonyx jubatus) Litters Using Different Vaccines. In Proceedings of the American Association of Zoo Veterinarians and European Association of Zoo and Wildlife Veterinarians Joint Conference, Toronto, ON, Canada, 4 October–5 November 2021. [Google Scholar]
- Scherba, G.; Hajjar, A.M.; Pernikoff, D.S.; Sundberg, J.P.; Basgall, E.J.; Leon-Monzon, M.; Nerurkar, L.; Reichmann, M.E. Comparison of a cheetah herpesvirus isolate to feline herpesvirus type 1. Arch. Virol. 1988, 100, 89–97. [Google Scholar] [CrossRef]
- Lewin, A.C.; Kolb, A.W.; McLellan, G.J.; Bentley, E.; Bernard, K.A.; Newbury, S.P.; Brandt, C.R. Genomic, Recombinational and Phylogenetic Characterization of Global Feline Herpesvirus 1 Isolates. Virology 2018, 518, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Vaz, P.K.; Job, N.; Horsington, J.; Ficorilli, N.; Studdert, M.J.; Hartley, C.A.; Gilkerson, J.R.; Browning, G.F.; Devlin, J.M. Low genetic diversity among historical and contemporary clinical isolates of felid herpesvirus 1. BMC Genom. 2016, 17, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, A.C.; Coghill, L.M.; Mironovich, M.; Liu, C.-C.; Carter, R.T.; Ledbetter, E.C. Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1. Viruses 2020, 12, 1421. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Grondin, B.; DeLuca, N. Herpes Simplex Virus Type 1 ICP4 Promotes Transcription Preinitiation Complex Formation by Enhancing the Binding of TFIID to DNA. J. Virol. 2000, 74, 11504–11510. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Curr. Protoc. Bioinform. 2003, 1, 6.5.1–6.5.14. [Google Scholar] [CrossRef]
- Kolb, A.W.; Lewin, A.C.; Moeller Trane, R.; McLellan, G.J.; Brandt, C.R. Phylogenetic and recombination analysis of the herpesvirus genus varicellovirus. BMC Genom. 2017, 18, 887. [Google Scholar] [CrossRef]
- Dohner, D.E.; Adams, S.G.; Gelb, L.D. Recombination in tissue culture between varicella-zoster virus strains. J. Med Virol. 1988, 24, 329–341. [Google Scholar] [CrossRef]
- Henderson, L.M.; Katz, J.B.; Erickson, G.A.; Mayfield, J.E. In vivo and in vitro genetic recombination between conventional and gene-deleted vaccine strains of pseudorabies virus. Am. J. Vet. Res. 1990, 51, 1656–1662. [Google Scholar] [PubMed]
- Kuny, C.V.; Bowen, C.D.; Renner, D.W.; Johnston, C.M.; Szpara, M.L. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol. 2020, 6, veaa013. [Google Scholar] [CrossRef] [PubMed]
- Rathbun, C.B.; Shipley, M.; Johnston, C.; Szpara, M. Tracking Herpes simplex virus 1 genomic diversity between non-familial, adult transmission partners with oral and genital infections. In Proceedings of the American Society for Virology 40th Annual Meeting, Online, 19–23 July 2021. [Google Scholar]
- Ross, M.G.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and measuring bias in sequence data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef] [Green Version]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Kundu, S.; Jensen, N.J.; Gray, E.R.; Jones, M.; Steinberg, S.; Gershon, A.; Kinchington, P.R.; Schmid, D.S.; Balloux, F.; et al. Deep sequencing of viral genomes provides insight into the evolution and pathogenesis of varicella zoster virus and its vaccine in humans. Mol. Biol. Evol. 2014, 31, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Markham, P.F.; Coppo, M.J.C.; Legione, A.R.; Markham, J.F.; Noormohammadi, A.H.; Browning, G.F.; Ficorilli, N.; Hartley, C.A.; Devlin, J.M. Attenuated Vaccines Can Recombine to Form Virulent Field Viruses. Science 2012, 337, 188. [Google Scholar] [CrossRef]
- Cohn, L.A. Feline Respiratory Disease Complex. Vet. Clin. N. Am. Small Anim. Pract. 2011, 41, 1273–1289. [Google Scholar] [CrossRef]
- Povey, R.C.; Johnson, R.H. Observations on the epidemiology and control of viral respiratory disease in cats. J. Small Anim. Pract. 1970, 11, 485–494. [Google Scholar] [CrossRef]
- Donaldson, A.I.; Ferris, N.P. The survival of some air-borne animal viruses in relation to relative humidity. Vet. Microbiol. 1976, 1, 413–420. [Google Scholar] [CrossRef]



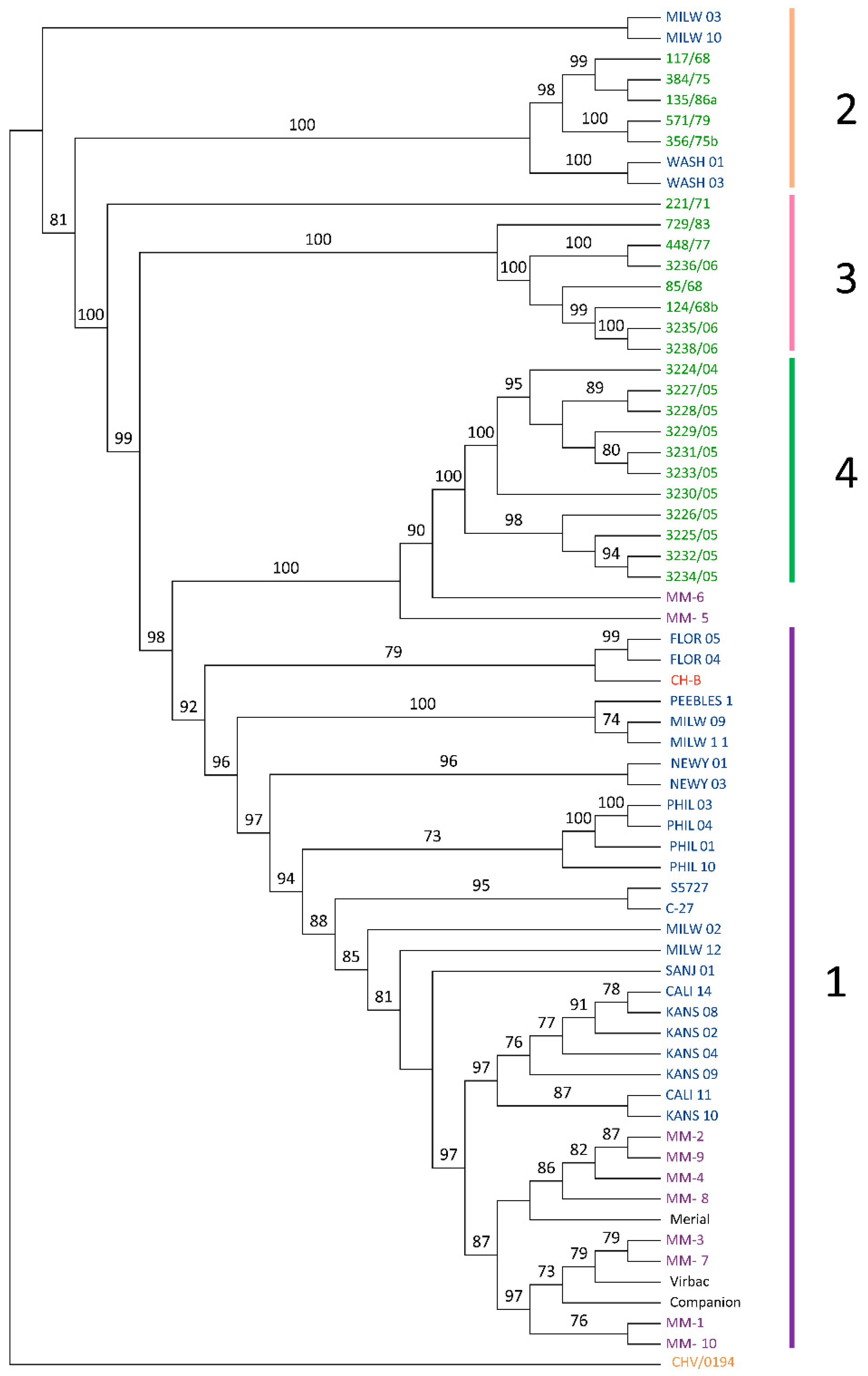{kind=link}
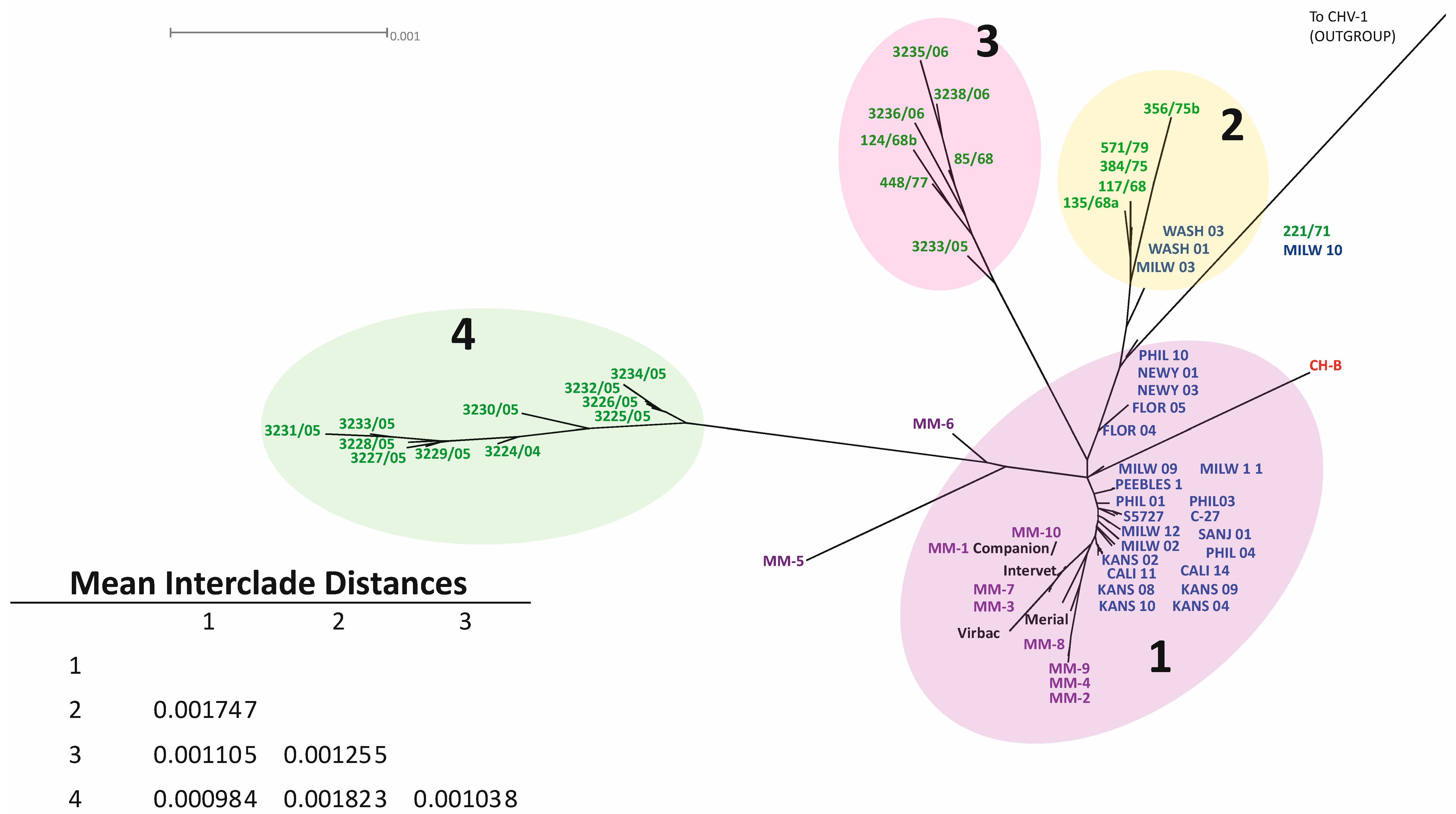{kind=link}
| Strain ID | GenBank Accession Number | Host Age (Years) | Host Sex | Host Species | Host Location | Sample Collection Date (Day/Month/Year) | Sample Source | Vaccine Status (Day/Month/Year) | Clinical Signs |
|---|---|---|---|---|---|---|---|---|---|
| MM-1 | OL321946 | Unknown | Unknown | Cheetah | Unknown | 2005 | Unknown | Unvaccinated | Unknown |
| MM-2 | OL410287 | 11 | Female | Cheetah | Florida | 16/12/2020 | Nasal swab | Merial MLV (16/12/2016) | None |
| MM-3 | OL410288 | 3.5 | Female | Cheetah | Florida | 16/12/2020 | Nasal swab | Merial MLV (20/7/2020) | Chronic dermatitis |
| MM-4 | OL410289 | 9 | Female | Cheetah | California | 3/6/2016 | Nasal swab | Unknown | Unknown |
| MM-5 | OL410290 | 6 | Male | Cheetah | Texas | 13/11/2013 | Nasal and pharyngeal swab | Unknown | Unknown |
| MM-6 | OL410291 | Unknown | Unknown | Cheetah | Missouri | 6/4/2001 | Nasal and ocular swab | Unknown | Unknown |
| MM-7 | OL410292 | 3.5 | Female | Cheetah | Florida | 16/12/2020 | Nasal swab | Merial MLV (20/7/2020) | Sneezing |
| MM-8 | OL410293 | 9 | Female | Cheetah | California | 1/6/2016 | Ocular swab | Unknown | Unknown |
| MM-9 | OL410294 | 7 | Female | Cheetah | Florida | 16/12/2020 | Nasal swab | Merial MLV (11/11/2020) | None |
| MM-10 | OL410295 | 0.75 | Male | Cheetah | Florida | 26/5/2009 | Nasal swab | Unknown | Productive cough and anorexia |
| Merial MLV | OL410296 |
| Strain | Number Reads | Mapped Reads | Average Mapped Read Length (bp) | Mean Coverage Per Base | GC Content (%) | Mapped Genome Length |
|---|---|---|---|---|---|---|
| MM-1 | 2,224,892 | 894,919 | 183.1 | 1199 | 45.9 | 137,392 |
| MM-2 | 2,596,572 | 724,887 | 183.0 | 970.2 | 45.8 | 137,037 |
| MM-3 | 2,438,790 | 790,548 | 183.6 | 1060.8 | 45.7 | 137,432 |
| MM-4 | 2,481,272 | 834,057 | 191.0 | 1167.1 | 45.3 | 136,996 |
| MM-5 | 2,200,776 | 609,757 | 179.6 | 801.6 | 45.2 | 136,712 |
| MM-6 | 2,537,734 | 832,788 | 190.9 | 1162.9 | 45.1 | 136,761 |
| MM-7 | 2,507,912 | 1,050,187 | 168.5 | 1294.6 | 45.7 | 137,611 |
| MM-8 | 2,106,004 | 693,291 | 188 | 955.2 | 45.3 | 136,818 |
| MM-9 | 1,983,946 | 640,857 | 178.6 | 836.2 | 45.8 | 136,966 |
| MM-10 | 2,199,852 | 520,743 | 191.4 | 729.8 | 45.3 | 136,658 |
| Merial MLV | 1,751,690 | 854,840 | 179.5 | 1122.2 | 45.9 | 137,390 |
| Gene | Number of Unique Variants (FHV-1 All Cheetah Isolates Compared with C-27) | Number of Unique Variants (MLV Strain Compared with Isolates from Vaccinated Cheetahs (MM-2, -3, -7, and -9)) | ||
|---|---|---|---|---|
| Synonymous Variants | Non-Synonymous Variants | Synonymous Variants | Non-Synonymous Variants | |
| ICP4 | 4 | 6 | 0 | 0 |
| UL10 | 0 | 1 | 0 | 0 |
| UL12 | 2 | 0 | 0 | 0 |
| UL16 | 0 | 2 | 0 | 0 |
| UL17 | 0 | 1 | 0 | 0 |
| UL19 | 1 | 0 | 0 | 0 |
| UL20 | 2 | 0 | 0 | 0 |
| UL25 | 1 | 2 | 0 | 0 |
| UL28 | 2 | 1 | 0 | 0 |
| UL29 | 0 | 1 | 0 | 0 |
| UL32 | 1 | 0 | 0 | 0 |
| UL36 | 3 | 0 | 0 | 0 |
| UL38 | 0 | 2 | 0 | 0 |
| UL4 | 1 | 0 | 0 | 0 |
| UL40 | 1 | 1 | 0 | 0 |
| UL41 | 2 | 3 | 0 | 1 |
| UL43 | 1 | 0 | 0 | 0 |
| UL44 | 0 | 1 | 0 | 0 |
| UL47 | 1 | 0 | 0 | 0 |
| UL48 | 0 | 1 | 0 | 4 |
| UL50 | 0 | 1 | 0 | 0 |
| UL52 | 0 | 1 | 0 | 0 |
| UL53 | 1 | 0 | 0 | 0 |
| UL54 | 1 | 1 | 0 | 0 |
| UL55 | 0 | 1 | 0 | 0 |
| UL7 | 0 | 1 | 0 | 0 |
| UL9 | 1 | 0 | 0 | 0 |
| US2 | 1 | 0 | 0 | 0 |
| US4 | 1 | 0 | 0 | 0 |
| US7 | 0 | 1 | 0 | 0 |
| US9 | 0 | 1 | 0 | 0 |
| UL3.5 | 1 | 1 | 0 | 0 |
| UL35 | 0 | 1 | 0 | 0 |
| UL5 | 0 | 1 | 0 | 0 |
| UL6 | 0 | 1 | 0 | 0 |
| Isolate | Percent Identity with Merial MLV | Percent Identity with Reference FHV-1 Isolate (C-27) |
|---|---|---|
| MM-1 | 99.03 | 98.31 |
| MM-2 | 99.04 | 98.32 |
| MM-3 | 99.08 | 98.38 |
| MM-4 | 98.88 | 98.17 |
| MM-5 | 98.62 | 98.05 |
| MM-6 | 98.70 | 98.23 |
| MM-7 | 98.86 | 98.43 |
| MM-8 | 99.04 | 98.35 |
| MM-9 | 99.01 | 98.39 |
| MM-10 | 99.05 | 98.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marino, M.E.; Mironovich, M.A.; Ineck, N.E.; Citino, S.B.; Emerson, J.A.; Maggs, D.J.; Coghill, L.M.; Dubovi, E.J.; Turner, R.C.; Carter, R.T.; et al. Full Viral Genome Sequencing and Phylogenomic Analysis of Feline Herpesvirus Type 1 (FHV-1) in Cheetahs (Acinonyx jubatus). Viruses 2021, 13, 2307. https://doi.org/10.3390/v13112307
Marino ME, Mironovich MA, Ineck NE, Citino SB, Emerson JA, Maggs DJ, Coghill LM, Dubovi EJ, Turner RC, Carter RT, et al. Full Viral Genome Sequencing and Phylogenomic Analysis of Feline Herpesvirus Type 1 (FHV-1) in Cheetahs (Acinonyx jubatus). Viruses. 2021; 13(11):2307. https://doi.org/10.3390/v13112307
Chicago/Turabian StyleMarino, Morgan E., Melanie A. Mironovich, Nikole E. Ineck, Scott B. Citino, Jessica A. Emerson, David J. Maggs, Lyndon M. Coghill, Edward J. Dubovi, Rachel C. Turner, Renee T. Carter, and et al. 2021. "Full Viral Genome Sequencing and Phylogenomic Analysis of Feline Herpesvirus Type 1 (FHV-1) in Cheetahs (Acinonyx jubatus)" Viruses 13, no. 11: 2307. https://doi.org/10.3390/v13112307
APA StyleMarino, M. E., Mironovich, M. A., Ineck, N. E., Citino, S. B., Emerson, J. A., Maggs, D. J., Coghill, L. M., Dubovi, E. J., Turner, R. C., Carter, R. T., & Lewin, A. C. (2021). Full Viral Genome Sequencing and Phylogenomic Analysis of Feline Herpesvirus Type 1 (FHV-1) in Cheetahs (Acinonyx jubatus). Viruses, 13(11), 2307. https://doi.org/10.3390/v13112307