
Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmid Construction
2.3. siRNA
2.4. Western Blotting
2.5. Immunofluorescent Staining
2.6. Analysis of Cell Cycle Profiles by CELAVIEW RS100
2.7. Analysis of Cell Cycle Profiles by Flow Cytometry
2.8. Immunoprecipitation
2.9. Protein Expression and Purification
2.10. Pull-Down Assay
2.11. Viral Stock and Viral Infection of Macrophages
2.12. Statistical Analysis
3. Results
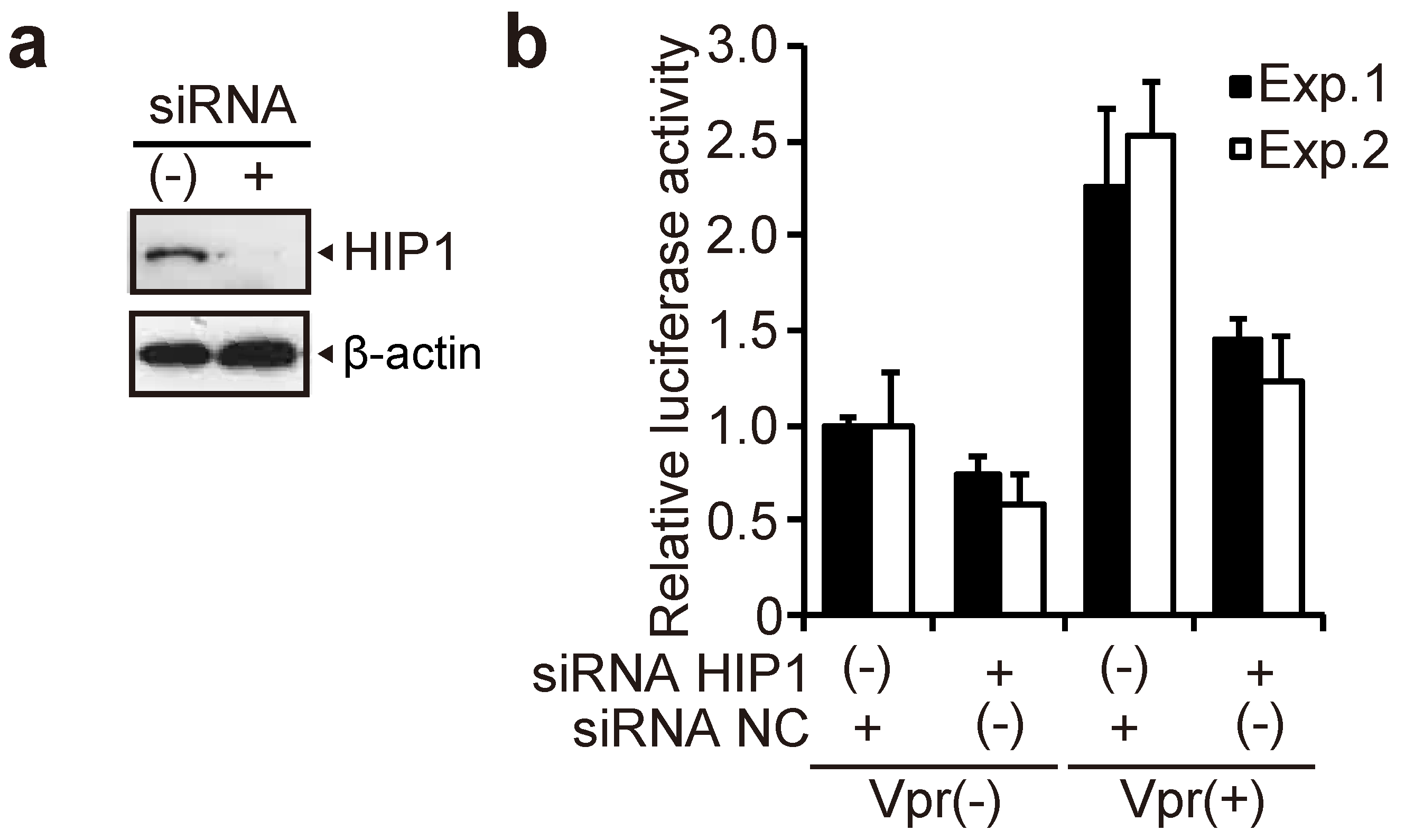
3.1. HIP1 Enhances Vpr-Induced G2 Arrest
3.2. HIP1 Interacts with Vpr
3.3. HIP1 Is Dispensable for Vpr-Induced DNA Double-Strand Breaks
3.4. HIP1 Promotes HIV-1 Infection in Macrophages
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hashizume, C.; Kuramitsu, M.; Zhang, X.; Kurosawa, T.; Kamata, M.; Aida, Y. Human immunodeficiency virus type 1 Vpr interacts with spliceosomal protein SAP145 to mediate cellular pre-mRNA splicing inhibition. Microbes Infect. 2007, 9, 490–497. [Google Scholar] [CrossRef]
- Kuramitsu, M.; Hashizume, C.; Yamamoto, N.; Azuma, A.; Kamata, M.; Yamamoto, N.; Tanaka, Y.; Aida, Y. A novel role for Vpr of human immunodeficiency virus type 1 as a regulator of the splicing of cellular pre-mRNA. Microbes Infect. 2005, 7, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Aida, Y. HIV-1 Vpr: A novel role in regulating RNA splicing. Curr. HIV Res. 2009, 7, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Chutiwitoonchai, N.; Siarot, L.; Takeda, E.; Shioda, T.; Ueda, M.; Aida, Y. HIV-1 Vpr Abrogates the Effect of TSG101 Overexpression to Support Virus Release. PLoS ONE 2016, 11, e0163100. [Google Scholar] [CrossRef] [Green Version]
- Kamata, M.; Nitahara-Kasahara, Y.; Miyamoto, Y.; Yoneda, Y.; Aida, Y. Importin-α Promotes Passage through the Nuclear Pore Complex of Human Immunodeficiency Virus Type 1 Vpr. J. Virol. 2005, 79, 3557–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitahara-Kasahara, Y.; Kamata, M.; Yamamoto, T.; Zhang, X.; Miyamoto, Y.; Muneta, K.; Iijima, S.; Yoneda, Y.; Tsunetsugu-Yokota, Y.; Aida, Y. Novel Nuclear Import of Vpr Promoted by Importin α Is Crucial for Human Immunodeficiency Virus Type 1 Replication in Macrophages. J. Virol. 2007, 81, 5284–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, S.; Rexach, M.; Zybarth, G.; Reiling, N.; Lee, M.; Ratner, L.; Lane, C.M.; Moore, M.S.; Blobel, G.; Bukrinsky, M. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 1998, 17, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr Overcomes Macrophage-Specific Restriction of HIV-1 Env Expression and Virion Production. Cell Host Microbe 2014, 16, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhou, T.; Frabutt, D.; Zheng, Y.-H. HIV-1 Vpr increases Env expression by preventing Env from endoplasmic reticulum-associated protein degradation (ERAD). Virology 2016, 496, 194–202. [Google Scholar] [CrossRef]
- Wang, Q.; Su, L. Vpr Enhances HIV-1 Env Processing and Virion Infectivity in Macrophages by Modulating TET2-Dependent IFITM3 Expression. mBio 2019, 10, e01344-19. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Wang, Q.; Xu, Y.; Tsao, L.-C.; Nakagawa, T.; Guo, H.; Su, L.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4VprBP E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961–970.e5. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Hao, C.; Shun, M.-C.; Kaur, S.; Swanson, S.K.; Florens, L.; Washburn, M.; Skowronski, J. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E3921–E3930. [Google Scholar] [CrossRef] [Green Version]
- Lahouassa, H.; Blondot, M.-L.; Chauveau, L.; Chougui, G.; Morel, M.; Leduc, M.; Guillonneau, F.; Ramirez, B.C.; Schwartz, O.; Margottin-Goguet, F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 5311–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Shun, M.-C.; Hao, C.; Zhang, Y.; Qian, J.; Hrecka, K.; DeLucia, M.; Monnie, C.; Ahn, J.; Skowronski, J. HIV-1 Vpr Reprograms CLR4 DCAF1 E3 Ubiquitin Ligase to Antagonize Exonuclease 1-Mediated Restriction of HIV-1 Infection. mBio 2018, 9, e01732-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Shun, M.-C.; Zhang, Y.; Hao, C.; Skowronski, J. HIV-1 Vpr counteracts HLTF-mediated restriction of HIV-1 infection in T cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9568–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Aida, Y. Visualizing Vpr-Induced G2 Arrest and Apoptosis. PLoS ONE 2014, 9, e86840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyavoo, V.; Mahboubi, A.; Mahalingam, S.; Ramalingam, R.; Kudchodkar, S.; Williams, W.V.; Green, D.R.; Weiner, D.B. HIV-1 Vpr suppresses immune activation and apoptosis through regulation of nuclear factor kappa B. Nat. Med. 1997, 3, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Jowett, J.B.; Planelles, V.; Poon, B.; Shah, N.P.; Chen, M.L.; Chen, I.S. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2+ M phase of the cell cycle. J. Virol. 1995, 69, 6304–6313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, M.; Kamata, M.; Mojin, T.; Nakai, Y.; Aida, Y. Induction of apoptosis by the Vpr protein of human immunodeficiency virus type 1 occurs independently of G(2) arrest of the cell cycle. Virology 2000, 276, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, M.; Hashimoto, Y.; Takeshima, S.-N.; Aida, Y. The human immunodeficiency virus type 1 Vpr protein and its carboxy-terminally truncated form induce apoptosis in tumor cells. Cancer Cell Int. 2009, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogel, M.E.; Wu, L.I.; Emerman, M.L. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Bieniasz, P.D. HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. eLife 2020, 9, e55806. [Google Scholar] [CrossRef]
- Chang, H.; Siarot, L.; Matsuura, R.; Lo, C.-W.; Sato, H.; Otsuki, H.; Aida, Y. Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins. Viruses 2020, 12, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.; Skowronski, J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguette, N.; Brégnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature Activation of the SLX4 Complex by Vpr Promotes G2/M Arrest and Escape from Innate Immune Sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, H.; Sanjoh, A.; Murakami, T.; Murakami, H.; Matsuda, G.; Hagiwara, K.; Yokoyama, M.; Sato, H.; Miyamoto, Y.; Dohmae, N.; et al. Molecular Mechanism of HIV-1 Vpr for Binding to Importin-α. J. Mol. Biol. 2016, 428, 2744–2757. [Google Scholar] [CrossRef]
- Murakami, H.; Suzuki, T.; Tsuchiya, K.; Gatanaga, H.; Taura, M.; Kudo, E.; Okada, S.; Takei, M.; Kuroda, K.; Yamamoto, T.; et al. Protein Arginine N-methyltransferases 5 and 7 Promote HIV-1 Production. Viruses 2020, 12, 355. [Google Scholar] [CrossRef] [Green Version]
- Planelles, V.; Jowett, J.B.; Li, Q.X.; Hahn, B.; Chen, I.S. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 1996, 70, 2516–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stivahtis, G.L.; Soares, M.A.; Vodicka, M.A.; Hahn, B.H.; Emerman, M. Conservation and host specificity of Vpr-mediated cell cycle arrest suggest a fundamental role in primate lentivirus evolution and biology. J. Virol. 1997, 71, 4331–4338. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Misawa, N.; Iwami, S.; Satou, Y.; Matsuoka, M.; Ishizaka, Y.; Ito, M.; Aihara, K.; An, D.S.; Koyanagi, Y. HIV-1 Vpr Accelerates Viral Replication during Acute Infection by Exploitation of Proliferating CD4+ T Cells In Vivo. PLoS Pathog. 2013, 9, e1003812. [Google Scholar] [CrossRef]
- Poon, B.; Jowett, J.B.; Stewart, S.A.; Armstrong, R.W.; Rishton, G.M.; Chen, I.S. Human immunodeficiency virus type 1 vpr gene induces phenotypic effects similar to those of the DNA alkylating agent, nitrogen mustard. J. Virol. 1997, 71, 3961–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, E.S.; Chen, J.; Andersen, J.L.; Ardon, O.; DeHart, J.L.; Blackett, J.; Choudhary, S.K.; Camerini, D.; Nghiem, P.; Planelles, V. Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol. Cell. Biol. 2004, 24, 9286–9294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, E.S.; Sherman, M.P.; Blackett, J.L.; Neidleman, J.A.; Kreis, C.; Mundt, P.; Williams, S.A.; Warmerdam, M.; Kahn, J.; Hecht, F.M.; et al. Human Immunodeficiency Virus Type 1 Vpr Induces DNA Replication Stress In Vitro and In Vivo. J. Virol. 2006, 80, 10407–10418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzile, J.-P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, É.A. HIV-1 Vpr-Mediated G2 Arrest Involves the DDB1-CUL4AVPRBP E3 Ubiquitin Ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Monteiro-Filho, C.M.; Argañaraz, E.R.; Planelles, V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 2007, 4, 57. [Google Scholar] [CrossRef] [Green Version]
- Le Rouzic, E.; Belaïdouni, N.; Estrabaud, E.; Morel, M.; Rain, J.-C.; Transy, C.; Margottin-Goguet, F. HIV1 Vpr Arrests the Cell Cycle by Recruiting DCAF1/VprBP, a Receptor of the Cul4-DDB1 Ubiquitin Ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Schröfelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 4130–4135. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Duus, K.M.; Friedrich, T.D.; de Noronha, C.M.C. The HIV1 Protein Vpr Acts to Promote G2 Cell Cycle Arrest by Engaging a DDB1 and Cullin4A-containing Ubiquitin Ligase Complex Using VprBP/DCAF1 as an Adaptor. J. Biol. Chem. 2007, 282, 27046–27057. [Google Scholar] [CrossRef] [Green Version]
- Berger, G.; Lawrence, M.; Hue, S.; Neil, S.J.D. G2/M Cell Cycle Arrest Correlates with Primate Lentiviral Vpr Interaction with the SLX4 Complex. J. Virol. 2014, 89, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Fregoso, O.I.; Emerman, M. Activation of the DNA Damage Response Is a Conserved Function of HIV-1 and HIV-2 Vpr That Is Independent of SLX4 Recruitment. mBio 2016, 7, e01433-16. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, J.M.; Fox, C.; Wahl, S.M. Macrophages as a Source of HIV During Opportunistic Infections. Science 1997, 276, 1857–1861. [Google Scholar] [CrossRef]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbramanian, R.A.; Kessous-Elbaz, A.; Lodge, R.; Forget, J.; Yao, X.-J.; Bergeron, D.; Cohen, E.A. Human Immunodeficiency Virus Type 1 Vpr Is a Positive Regulator of Viral Transcription and Infectivity in Primary Human Macrophages. J. Exp. Med. 1998, 187, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Kalchman, M.A.; Koide, H.B.; McCutcheon, K.; Graham, R.K.; Nichol, K.; Nishiyama, K.; Kazemi-Esfarjani, P.; Lynn, F.C.; Wellington, C.; Metzler, M.; et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat. Genet. 1997, 16, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, N.; Peoples, R.; Himmelbauer, H.; Wanker, E.E.; Lehrach, H.; Francke, U. Localization of the HumanHIP1Gene Close to the Elastin (ELN) Locus on 7q11.23. Genomics 1997, 46, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Legendre-Guillemin, V.; Gan, L.; Chopra, V.; Kwok, A.; McPherson, P.S.; Hayden, M. HIP1 Functions in Clathrin-mediated Endocytosis through Binding to Clathrin and Adaptor Protein 2. J. Biol. Chem. 2001, 276, 39271–39276. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Agostinelli, N.R.; Brett, T.J.; Mizukami, I.; Ross, T.; Traub, L. Clathrin- and AP-2-binding Sites in HIP1 Uncover a General Assembly Role for Endocytic Accessory Proteins. J. Biol. Chem. 2001, 276, 46230–46236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waelter, S.; Scherzinger, E.; Hasenbank, R.; Nordhoff, E.; Lurz, R.; Goehler, H.; Gauss, C.; Sathasivam, K.; Bates, G.P.; Lehrach, H.; et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum. Mol. Genet. 2001, 10, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Rao, D.S.; Bradley, S.; Kumar, P.D.; Hyun, T.; Saint-Dic, D.; Oravecz-Wilson, K.; Kleer, C.G.; Ross, T.S. Altered receptor trafficking in Huntingtin Interacting Protein 1-transformed cells. Cancer Cell 2003, 3, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Rao, D.S.; Hyun, T.S.; Kumar, P.D.; Mizukami, I.F.; Rubin, M.A.; Lucas, P.C.; Sanda, M.G.; Ross, T.S. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J. Clin. Investig. 2002, 110, 351–360. [Google Scholar] [CrossRef]
- Choi, S.A.; Kim, S.J.; Chung, K.C. Huntingtin-interacting protein 1-mediated neuronal cell death occurs through intrinsic apoptotic pathways and mitochondrial alterations. FEBS Lett. 2006, 580, 5275–5282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.E.; Choi, S.A.; Park, J.B.; Chung, K.C. Regulation of the proapoptotic activity of huntingtin interacting protein 1 by Dyrk1 and caspase-3 in hippocampal neuroprogenitor cells. J. Neurosci. Res. 2005, 81, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Myojin, T.; Kamata, M.; Aida, Y. Human Immunodeficiency Virus Type 1VprGene Product Prevents Cell Proliferation on Mouse NIH3T3 Cells without the G2Arrest of the Cell Cycle. Biochem. Biophys. Res. Commun. 1997, 232, 550–554. [Google Scholar] [CrossRef]
- Takeda, E.; Murakami, T.; Matsuda, G.; Murakami, H.; Zako, T.; Maeda, M.; Aida, Y. Nuclear Exportin Receptor CAS Regulates the NPI-1–Mediated Nuclear Import of HIV-1 Vpr. PLoS ONE 2011, 6, e27815. [Google Scholar] [CrossRef] [Green Version]
- Kameoka, M.; Kitagawa, Y.; Utachee, P.; Jinnopat, P.; Dhepakson, P.; Isarangkura-Na-Ayuthaya, P.; Tokunaga, K.; Sato, H.; Komano, J.; Yamamoto, N.; et al. Identification of the suppressive factors for human immunodeficiency virus type-1 replication using the siRNA mini-library directed against host cellular genes. Biochem. Biophys. Res. Commun. 2007, 359, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.-P.; Abrahamyan, L.G.; Gerard, F.C.A.; Rougeau, N.; Cohen, É.A. Formation of Mobile Chromatin-Associated Nuclear Foci Containing HIV-1 Vpr and VPRBP Is Critical for the Induction of G2 Cell Cycle Arrest. PLoS Pathog. 2010, 6, e1001080. [Google Scholar] [CrossRef] [Green Version]
- Belzile, J.-P.; Richard, J.; Rougeau, N.; Xiao, Y.; Cohen, E.A. HIV-1 Vpr Induces the K48-Linked Polyubiquitination and Proteasomal Degradation of Target Cellular Proteins To Activate ATR and Promote G 2 Arrest. J. Virol. 2010, 84, 3320–3330. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nat. Cell Biol. 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Elder, R.T.; Qin, K.; Park, H.U.; Liang, D.; Zhao, R.Y. Phosphatase Type 2A-dependent and -independent Pathways for ATR Phosphorylation of Chk1. J. Biol. Chem. 2007, 282, 7287–7298. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Lopez, A.; Sandoval, C.; Doyle, R.N.; Fregoso, O.I. HIV Vpr Modulates the Host DNA Damage Response at Two Independent Steps to Damage DNA and Repress Double-Strand DNA Break Repair. mBio 2020, 11, e00940-20. [Google Scholar] [CrossRef]
- Nishizawa, M.; Kamata, M.; Katsumata, R.; Aida, Y. A Carboxy-Terminally Truncated Form of the Human Immunodeficiency Virus Type 1 Vpr Protein Induces Apoptosis via G 1 Cell Cycle Arrest. J. Virol. 2000, 6058–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senetar, M.A.; Foster, S.J.; McCann, R.O. Intrasteric Inhibition Mediates the Interaction of the I/LWEQ Module Proteins Talin1, Talin2, Hip1, and Hip12 with Actin. Biochemistry 2004, 43, 15418–15428. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV–human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4+ T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, D.L.; Lenardo, M.J. Vpr Cytopathicity Independent of G2/M Cell Cycle Arrest in Human Immunodeficiency Virus Type 1-Infected CD4+ T Cells. J. Virol. 2007, 81, 8878–8890. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, T.; Matsuura, R.; Chutiwitoonchai, N.; Takei, M.; Aida, Y. Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages. Viruses 2021, 13, 2308. https://doi.org/10.3390/v13112308
Murakami T, Matsuura R, Chutiwitoonchai N, Takei M, Aida Y. Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages. Viruses. 2021; 13(11):2308. https://doi.org/10.3390/v13112308
Chicago/Turabian StyleMurakami, Tomoyuki, Ryosuke Matsuura, Nopporn Chutiwitoonchai, Masami Takei, and Yoko Aida. 2021. "Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages" Viruses 13, no. 11: 2308. https://doi.org/10.3390/v13112308
APA StyleMurakami, T., Matsuura, R., Chutiwitoonchai, N., Takei, M., & Aida, Y. (2021). Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages. Viruses, 13(11), 2308. https://doi.org/10.3390/v13112308