
The Roles of Neutrophils in Cytokine Storms

 , , ,
, , ,  ,
,  , ,
, ,  and
and 
{kind=link}
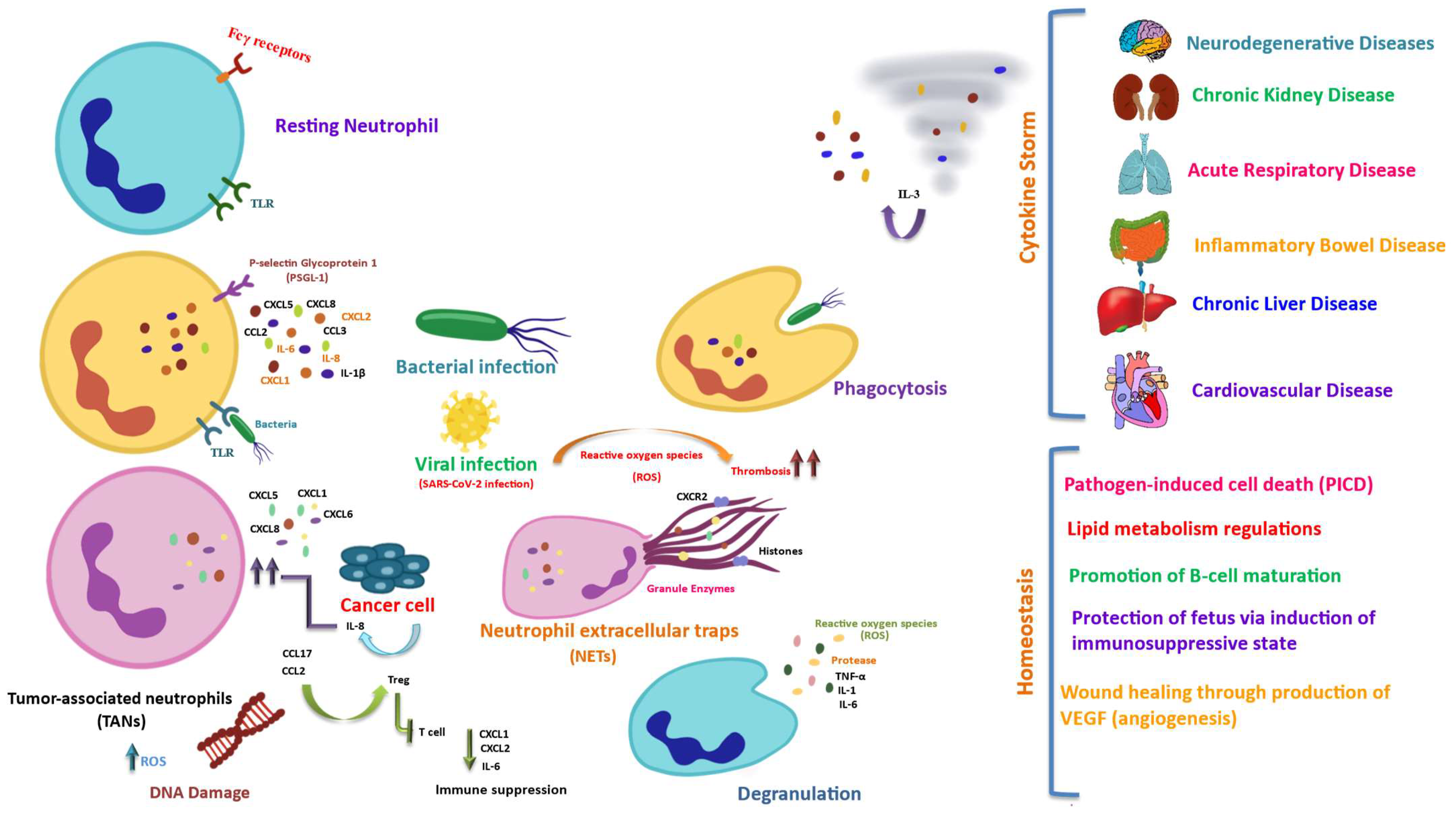
Abstract
:1. Introduction
2. Immunobiology of Neutrophils and Neutrophil-Derived Cytokines
3. Neutrophil Hyperactivation and CS
3.1. Viral Infections
3.1.1. RSV
3.1.2. Influenza Virus
3.1.3. Highly Pathogenic Coronaviruses
3.2. Bacterial Infection
3.3. Cancers
3.4. Autoimmunity
3.4.1. Multiple Sclerosis (MS)
3.4.2. Rheumatoid Arthritis (RA)
3.4.3. Systemic Lupus Erythematosus (SLE)
3.5. Neutrophils as a Therapeutic Target
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrara, J.L.; Abhyankar, S.; Gilliland, D.G. Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transplant. Proc. 1993, 25, 1216–1217. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.; Alves, J.D. Pathogenesis of multi-organic failure in autoimmune diseases. Autoimmun. Rev. 2009, 8, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Murthy, H.; Iqbal, M.; Chavez, J.C.; A Kharfan-Dabaja, M. Cytokine release syndrome: Current perspectives. ImmunoTargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Jeffrey, F.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Porter, D. Cytokine release syndrome with chimeric antigen receptor T Cell therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, L.E.; Leppkes, M.; Fuchs, T.A.; Hoffmann, M.; Herrmann, M. Missing in action—The meaning of cell death in tissue damage and inflammation. Immunol. Rev. 2017, 280, 26–40. [Google Scholar] [CrossRef]
- Karbian, N.; Abutbul, A.; El-Amore, R.; Eliaz, R.; Beeri, R.; Reicher, B.; Mevorach, D. Apoptotic cell therapy for cytokine storm associated with acute severe sepsis. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The immunology of macrophage activation syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Perez, N.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Fischer, A.; Griscelli, C. Impaired natural killer activity in lymphohistiocytosis syndrome. J. Pediatr. 1984, 104, 569–573. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoller, E.E.; Lykens, J.E.; Terrell, C.E.; Aliberti, J.; Filipovich, A.H.; Henson, P.M.; Jordan, M.B. Hemophagocytosis causes a consumptive anemia of inflammation. J. Exp. Med. 2011, 208, 1203–1214. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Unger, S.A.; Walton, M.; Schwarze, J. The human immune response to respiratory syncytial virus infection. Clin. Microbiol. Rev. 2017, 30, 481–502. [Google Scholar] [CrossRef] [Green Version]
- Rungelrath, V.; Kobayashi, S.D.; DeLeo, F.R. Neutrophils in innate immunity and systems biology-level approaches. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1458. [Google Scholar] [CrossRef]
- KA, A.-A.; Al-Anazi, W.; Al-Jasser, A. Neutrophils, NETs, NETosis and their paradoxical roles in COVID-19. J. Stem. Cell Ther. Transplant. 2020, 4, 3–10. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A review of neutrophil extracellular traps (NETs) in disease: Potential anti-NETs therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef] [PubMed]
- De Buhr, N.; Von Köckritz-Blickwede, M. How neutrophil extracellular traps become visible. J. Immunol. Res. 2016, 2016, 4604713. [Google Scholar] [CrossRef] [Green Version]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef]
- Cassatella, M.A.; Östberg, N.; Tamassia, N.; Soehnlein, O. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol. 2019, 40, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; Nicholls, A.J.; Wong, C.H. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Bouchery, T.; Harris, N. Neutrophil–macrophage cooperation and its impact on tissue repair. Immunol. Cell Biol. 2019, 97, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Puga, I.; Cols, M.; Barra, C.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S. Influenza-associated encephalopathy--pathophysiology and disease mechanisms. Nihon rinsho. Jpn. J. Clin. Med. 2003, 61, 1953–1958. [Google Scholar]
- Barry, S.M.; A Johnson, M.; Janossy, G. Cytopathology or immunopathology? The puzzle of cytomegalovirus pneumonitis revisited. Bone Marrow Transplant. 2000, 26, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Jahrling, P.B.; Hensley, L.E.; Martinez, M.J.; LeDuc, J.W.; Rubins, K.H. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl. Acad. Sci. USA 2004, 101, 15196–15200. [Google Scholar] [CrossRef] [Green Version]
- Imashuku, S. Clinical features and treatment strategies of Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis. Crit. Rev. Oncol. 2002, 44, 259–272. [Google Scholar] [CrossRef]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-γ-related cytokine storm in SARS patients. J. Med Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R. Cytokine storms in infectious diseases. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 501–503. [Google Scholar] [CrossRef] [Green Version]
- Naumenko, V.; Turk, M.; Jenne, C.N.; Kim, S.-J. Neutrophils in viral infection. Cell Tissue Res. 2018, 371, 505–516. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.; de Graaff, P.M.; Kimpen, J.L.; Koenderman, L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-κB-dependent mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar] [CrossRef] [Green Version]
- Rawal, A.; Gavin, P.J.; Sturgis, C.D. Cerebrospinal fluid cytology in seasonal epidemic West Nile virus meningoencephalitis. Diagn. Cytopathol. 2006, 34, 127–129. [Google Scholar] [CrossRef]
- Bai, F.; Kong, K.-F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgometry, R.R. A paradoxical role for neutrophils in the pathogenesis of west nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- Grundy, J.E.; Lawson, K.M.; MacCormac, L.P.; Fletcher, J.; Yong, K.L. Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J. Infect. Dis. 1998, 177, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Savard, M.; Gosselin, J. Epstein-Barr virus immunossuppression of innate immunity mediated by phagocytes. Virus Res. 2006, 119, 134–145. [Google Scholar] [CrossRef]
- Anderson, L.J.; Heilman, C.A. Protective and disease-enhancing immune responses to respiratory syncytial virus. J. Infect. Dis. 1995, 171, 1–7. [Google Scholar] [CrossRef]
- Everard, M.L.; Swarbrick, A.; Wrightham, M.; McIntyre, J.; Dunkley, C.; James, P.D.; Sewell, H.F.; Milner, A.D. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch. Dis. Child. 1994, 71, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Wang, S.Z.; Dowling, K.D.; Forsyth, K.D. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J. Paediatr. Child Health 2001, 37, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, M.J.; Openshaw, P.J.; Askonas, B.A. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 1988, 168, 1163–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, D.R.; Ansar, M.; Ivanciuc, T.; Qu, Y.; Casola, A.; Garofalo, R.P. Selective blockade of TNFR1 improves clinical disease and bronchoconstriction in experimental RSV infection. Viruses 2020, 12, 1176. [Google Scholar] [CrossRef] [PubMed]
- Eash, K.J.; Means, J.M.; White, D.; Link, D.C. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 2009, 113, 4711–4719. [Google Scholar] [CrossRef] [Green Version]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofidou-Solomidou, M.; Nakada, M.T.; Williams, J.; A Muller, W.; Delisser, H.M. Neutrophil platelet endothelial cell adhesion molecule-1 participates in neutrophil recruitment at inflammatory sites and is down-regulated after leukocyte extravasation. J. Immunol. 1997, 158, 4872–4878. [Google Scholar] [PubMed]
- Atsuta, J.; Sterbinsky, S.A.; Plitt, J.; Schwiebert, L.M.; Bochner, B.S.; Schleimer, R.P. Phenotyping and cytokine regulation of the BEAS-2B human bronchial epithelial cell: Demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am. J. Respir. Cell Mol. Biol. 1997, 17, 571–582. [Google Scholar] [CrossRef]
- Wang, S.-Z.; Forsyth, K.D. The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 2000, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-Z.; Smith, P.K.; Lovejoy, M.; Bowden, J.; Alpers, J.H.; Forsyth, K. Shedding of L-selectin and PECAM-1 and upregulation of Mac-1 and ICAM-1 on neutrophils in RSV bronchiolitis. Am. J. Physiol. Cell. Mol. Physiol. 1998, 275, L983–L989. [Google Scholar] [CrossRef]
- Wang, S.Z.; Xu, H.; Wraith, A.; Bowden, J.J.; Alpers, J.H.; Forsyth, K.D. Neutrophils induce damage to respiratory epithelial cells infected with respiratory syncytial virus. Eur. Respir. J. 1998, 12, 612–618. [Google Scholar] [CrossRef] [Green Version]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.; Stein, R.; Bonorino, C.B.C.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4–dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Pennycook, A.; Openshaw, P.J.M. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef]
- Wang, S.Z.; Smith, P.K.; Lovejoy, M.; Bowden, J.; Alpers, J.H.; Forsyth, K. The apoptosis of neutrophils is accelerated in respiratory syncytial virus (RSV)-induced bronchiolitis. Clin. Exp. Immunol. 1998, 114, 49–54. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus ns1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, I.; Smith, G.; Ruf-Zamojski, F.; Martínez-Romero, C.; Fribourg, M.; Carbajal, E.A.; Hartmann, B.; Nair, V.D.; Marjanovic, N.; Monteagudo, P.L.; et al. Innate immune response to influenza virus at single-cell resolution in human epithelial cells revealed paracrine induction of interferon lambda 1. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Matsukura, S.; Kokubu, F.; Kubo, H.; Tomita, T.; Tokunaga, H.; Kadokura, M.; Yamamoto, T.; Kuroiwa, Y.; Ohno, T.; Suzaki, H.; et al. Expression of RANTES by normal airway epithelial cells after influenza virus A infection. Am. J. Respir. Cell Mol. Biol. 1998, 18, 255–264. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.; Ioannidis, L.J.; Croker, B.; Brown, L.; Brooks, A.; Reading, P.C. The role of neutrophils during mild and severe influenza virus infections of mice. PLoS ONE 2011, 6, e17618. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Wareing, M.D.; Shea, A.L.; Inglis, C.A.; Dias, P.B.; Sarawar, S.R. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 2007, 20, 369–378. [Google Scholar] [CrossRef]
- Tripathi, S.; Wang, G.; White, M.; Qi, L.; Taubenberger, J.; Hartshorn, K.L. Antiviral activity of the human cathelicidin, LL-37, and derived peptides on seasonal and pandemic influenza A viruses. PLoS ONE 2015, 10, e0124706. [Google Scholar] [CrossRef] [Green Version]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef]
- Malachowa, N.; Freedman, B.; Sturdevant, D.E.; Kobayashi, S.D.; Nair, V.; Feldmann, F.; Starr, T.; Steele-Mortimer, O.; Kash, J.C.; Taubenberger, J.K.; et al. Differential ability of pandemic and seasonal H1N1 influenza a viruses to alter the function of human neutrophils. mSphere 2018, 3, e00567-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.L.Y.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L.; Chan, M.C.W.; Chan, R.W.Y. Host DNA released by NETosis in neutrophils exposed to seasonal H1N1 and highly pathogenic H5N1 influenza viruses. Respir. Res. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.; Tripathi, S.; White, M.; Qi, L.; Taubenberger, J.; van Eijk, M.; Haagsman, H.; Hartshorn, K.L. Arginine-rich histones have strong antiviral activity for influenza A viruses. Innate Immun. 2015, 21, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J.G. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLOS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.A.; Plowden, J.K.; Garcia-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLOS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufford, M.M.; Richardson, G.; Zhou, H.; Manicassamy, B.; García-Sastre, A.; Enelow, R.I.; Braciale, T.J. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8+ T cells. PLoS ONE 2012, 7, e46581. [Google Scholar] [CrossRef]
- Berri, F.; Lê, V.B.; Jandrot-Perrus, M.; Lina, B.; Riteau, B. Switch from protective to adverse inflammation during influenza: Viral determinants and hemostasis are caught as culprits. Cell. Mol. Life Sci. 2014, 71, 885–898. [Google Scholar] [CrossRef]
- Kumar, Y.; Liang, C.; Limmon, G.V.; Liang, L.; Engelward, B.P.; Ooi, E.E.; Chen, J.; Tannenbaum, S.R. Molecular analysis of serum and bronchoalveolar lavage in a mouse model of influenza reveals markers of disease severity that can be clinically useful in humans. PLoS ONE 2014, 9, e86912. [Google Scholar] [CrossRef]
- Marion, T.; Elbahesh, H.; Thomas, P.G.; DeVincenzo, J.P.; Webby, R.; Schughart, K. Respiratory mucosal proteome quantification in human influenza infections. PLoS ONE 2016, 11, e0153674. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Fang, P.; He, R.; Li, M.; Yu, H.; Zhou, L.; Yi, Y.; Wang, F.; Rong, Y.; Zhang, Y.; et al. O-GlcNAc transferase promotes influenza A virus–induced cytokine storm by targeting interferon regulatory factor–5. Sci. Adv. 2020, 6, eaaz7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [CrossRef]
- Didangelos, A. COVID-19 hyperinflammation: What about neutrophils? MSphere 2020, 5, e00367-20. [Google Scholar] [CrossRef]
- Miller, E.J.; Cohen, A.B.; Nagao, S.; Griffith, D.; Maunder, R.J.; Martin, T.R.; Weiner-Kronish, J.P.; Sticherling, M.; Christophers, E.; Matthay, M.A. Elevated levels of NAP-1/Interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am. Rev. Respir. Dis. 1992, 146, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 2020, 27, 883–890. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. BioRxiv 2020, 2, 99–112. [Google Scholar] [CrossRef]
- Ma, L.; Sahu, S.K.; Cano, M.; Kuppuswamy, V.; Bajwa, J.; Pine, A.; Meizlish, M.; Goshua, G.; Chang, C.-H.; Zhang, H.; et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabh2259. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.-H.; Bang, J.H.; et al. Clinical progression and cytokine profiles of middle east respiratory syndrome coronavirus infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect. 2020, 81, e6–e12. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Mittal, S.; Gollapudi, S.; Butzmann, A.; Kumar, J.; Ohgami, R.S. A meta-analysis of SARS-CoV-2 patients identifies the combinatorial significance of D-dimer, C-reactive protein, lymphocyte, and neutrophil values as a predictor of disease severity. Int. J. Lab. Hematol. 2021, 43, 324–328. [Google Scholar] [CrossRef]
- Tsui, P.T.; Kwok, M.L.; Yuen, H.; Lai, S.T. Severe acute respiratory syndrome: Clinical outcome and prognostic correlates. Emerg. Infect. Dis. 2003, 9, 1064–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, C.-K.; Cheon, S.; Ha, N.-Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.-Y.; Inn, K.-S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef]
- Fu, J.; Kong, J.; Wang, W.; Wu, M.; Yao, L.; Wang, Z.; Jin, J.; Wu, D.; Yu, X. The clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retrospective study in Suzhou China. Thromb. Res. 2020, 192, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tatum, D.; Taghavi, S.; Houghton, A.; Stover, J.; Toraih, E.; Duchesne, J. Neutrophil-to-lymphocyte ratio and outcomes in louisiana COVID-19 patients. Shock 2020, 54, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-P.; Liu, J.-P.; Tao, W.-Q.; Li, H.-M. The diagnostic and predictive role of NLR, d-NLR and PLR in COVID-19 patients. Int. Immunopharmacol. 2020, 84, 106504. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Mo, P.; Liu, J.; Wang, H.; Wang, F.; Zhao, Q. Neutrophil to CD4+ lymphocyte ratio as a potential biomarker in predicting virus negative conversion time in COVID-19. Int. Immunopharmacol. 2020, 85, 106683. [Google Scholar] [CrossRef]
- Varim, C.; Yaylaci, S.; Demirci, T.; Kaya, T.; Nalbant, A.; Dheir, H.; Senocak, D.; Kurt, R.; Cengiz, H.; Karacaer, C. Neutrophil count to albumin ratio as a new predictor of mortality in patients with COVID-19 ınfection. Rev. Assoc. Méd. Bras. 2020, 66, 77–81. [Google Scholar] [CrossRef]
- Meizlish, M.L.; Pine, A.B.; Bishai, J.D.; Goshua, G.; Nadelmann, E.R.; Simonov, M.; Chang, C.H.; Zhang, H.; Shallow, M.; Bahel, P.; et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021, 5, 1164–1177. [Google Scholar] [CrossRef]
- Parackova, Z.; Zentsova, I.; Bloomfield, M.; Vrabcova, P.; Smetanova, J.; Klocperk, A.; Mesežnikov, G.; Mendez, L.F.C.; Vymazal, T.; Sediva, A. Disharmonic inflammatory signatures in COVID-19: Augmented neutrophils’ but impaired monocytes’ and dendritic cells’ responsiveness. Cells 2020, 9, 2206. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 2020, 182, 1419–1440. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.J.; Schroder, K. Pattern recognition receptor function in neutrophils. Trends Immunol. 2013, 34, 317–328. [Google Scholar] [CrossRef]
- Johansson, C.; Kirsebom, F.C.M. Neutrophils in respiratory viral infections. Mucosal Immunol. 2021, 14, 1–13. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453. [Google Scholar] [CrossRef]
- Hemmat, N.; Derakhshani, A.; Baghi, H.B.; Silvestris, N.; Baradaran, B.; De Summa, S. Neutrophils, crucial, or harmful immune cells involved in coronavirus infection: A bioinformatics study. Front. Genet. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.-C.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Yang, H.; Lu, Y.; Zhu, G.; Yang, L.; Zhao, Y.; Hu, B.; Ying, T. Interleukin 8 targeted contrast echocardiography is effective to evaluate myocardial ischemia-reperfusion injury in the rabbits. Biomed. Pharmacother. 2019, 109, 1346–1350. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, E.A.; Bersch, K.L.; Lauro, M.; Grimes, C.L. Differential peptidoglycan recognition assay using varied surface presentations. J. Am. Chem. Soc. 2020, 142, 10926–10930. [Google Scholar] [CrossRef] [PubMed]
- Demaret, J.; Venet, F.; Friggeri, A.; Cazalis, M.-A.; Plassais, J.; Jallades, L.; Malcus, C.; Poitevin-Later, F.; Textoris, J.; Lepape, A.; et al. Marked alterations of neutrophil functions during sepsis-induced immunosuppression. J. Leukoc. Biol. 2015, 98, 1081–1090. [Google Scholar] [CrossRef]
- Surewaard, B.G.; Trzciński, K.; Jacobino, S.R.; Hansen, I.S.; Vughs, M.M.; Sanders, E.A.; van der Ende, A.; van Strijp, J.A.; De Haas, C.J. Pneumococcal immune evasion: ZmpC inhibits neutrophil influx. Cell. Microbiol. 2013, 15, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Berends, E.T.M.; Chan, R.; Schwab, E.; Roy, S.; Sen, C.K.; Torres, V.; Wozniak, D.J. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl. Acad. Sci. USA 2018, 115, 7416–7421. [Google Scholar] [CrossRef] [Green Version]
- Chousterman, B.G.; Swirski, F.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 517–528. [Google Scholar] [CrossRef]
- Weber, G.F.; Chousterman, B.G.; He, S.; Fenn, A.M.; Nairz, M.; Anzai, A.; Brenner, T.; Uhle, F.; Iwamoto, Y.; Robbins, C.S.; et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 2015, 347, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Bhatty, M.; Tan, W.; Basco, M.; Pruett, S.; Nanduri, B. Binge alcohol consumption 18 h after induction of sepsis in a mouse model causes rapid overgrowth of bacteria, a cytokine storm, and decreased survival. Alcohol 2017, 63, 9–17. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: Apopulation-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
- Simon, T.G.; Roelstraete, B.; Sharma, R.; Khalili, H.; Hagström, H.; Ludvigsson, J.F. Cancer risk in patients with biopsy-confirmed nonalcoholic fatty liver disease: A population-based cohort study. Hepatology 2021, 74, 2410–2423. [Google Scholar] [CrossRef]
- Su, F.-H.; Bai, C.-H.; Le, T.N.; Muo, C.-H.; Chang, S.-N.; Te, A.; Sung, F.-C.; Yeh, C.-C. Patients with chronic hepatitis c virus infection are at an increased risk of colorectal cancer: A nationwide population-based case-control study in Taiwan. Front. Oncol. 2021, 10, 3002. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; Van Miltenburg, M.H.; Slagter, M.; De Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.-S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Cupp, M.A.; Cariolou, M.; Tzoulaki, I.; Aune, D.; Evangelou, E.; Berlanga-Taylor, A.J. Neutrophil to lymphocyte ratio and cancer prognosis: An umbrella review of systematic reviews and meta-analyses of observational studies. BMC Med. 2020, 18, 360. [Google Scholar] [CrossRef] [PubMed]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.-P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Nishida, J.; Momoi, Y.; Miyakuni, K.; Tamura, Y.; Takahashi, K.; Koinuma, D.; Miyazono, K.; Ehata, S. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat. Cell Biol. 2020, 22, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Braster, R.; O’Toole, T.; van Egmond, M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods 2014, 65, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.T.; Khan, H.Y.; Ahmad, A.; Weston, L.L.; A Nofchissey, R.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013, 62, 1745–1756. [Google Scholar] [CrossRef]
- Sawanobori, Y.; Ueha, S.; Kurachi, M.; Shimaoka, T.; Talmadge, J.E.; Abe, J.; Shono, Y.; Kitabatake, M.; Kakimi, K.; Mukaida, N.; et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood J. Am. Soc. Hematol. 2008, 111, 5457–5466. [Google Scholar] [CrossRef] [Green Version]
- Van Raam, B.J.; Drewniak, A.; Groenewold, V.; van den Berg, T.K.; Kuijpers, T.W. Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood J. Am. Soc. Hematol. 2008, 112, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016, 150, 1646–1658. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Rivera, C.; Khaznadar, S.S.; Shwin, K.W.; Irizarry-Caro, J.A.; O’Neil, L.J.; Liu, Y.; Jacobson, K.A.; Ombrello, A.; Stone, D.L.; Tsai, W.L.; et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019, 134, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, J. Neutrophil Extracellular Traps: A New Player in Cancer Metastasis and Therapeutic Target. J. Exp. Clin. Cancer Res. 2021, 40, 233. [Google Scholar] [CrossRef]
- Elliott, M.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovitch, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nat. Cell Biol. 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.; Darcy, P.; Smyth, M.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Ko, S.Y.; Mohamed, M.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Karschnia, P.; Blobner, J.; Teske, N.; Schöberl, F.; Fitzinger, E.; Dreyling, M.; Tonn, J.-C.; Thon, N.; Subklewe, M.; von Baumgarten, L. CAR T-Cells for CNS Lymphoma: Driving into New Terrain? Cancers 2021, 13, 2503. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Knezevic, A.; Pennisi, M.; Chen, Y.; Ruiz, J.D.; Purdon, T.J.; Devlin, S.M.; Smith, M.; Shah, G.L.; Halton, E.; et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020, 4, 3776–3787. [Google Scholar] [CrossRef]
- Dunleavy, K.; Hakim, F.; Kim, H.K.; Janik, J.E.; Grant, N.; Nakayama, T.; White, T.; Wright, G.; Kwak, L.; Gress, R.; et al. B-cell recovery following rituximab-based therapy is associated with perturbations in stromal derived factor-1 and granulocyte homeostasis. Blood 2005, 106, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Moradi, E.V.; Teimouri, A.; Rezaee, R.; Morovatdar, N.; Foroughian, M.; Layegh, P.; Kakhki, B.R.; Koupaei, S.R.A.; Ghorani, V. Increased age, neutrophil-to-lymphocyte ratio (NLR) and white blood cells count are associated with higher COVID-19 mortality. Am. J. Emerg. Med. 2021, 40, 11–14. [Google Scholar] [CrossRef]
- Lekstrom-Himes, J.A.; Gallin, J.I. Immunodeficiency diseases caused by defects in phagocytes. N. Engl. J. Med. 2000, 343, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Mócsai, A. The role of neutrophils in autoimmune diseases. Immunol. Lett. 2012, 143, 9–19. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Pham, C.T.N. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Thieblemont, N.; Wright, H.L.; Edwards, S.W.; Witko-Sarsat, V. Human neutrophils in auto-immunity. In Seminars in Immunopathology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 159–173. [Google Scholar]
- Kaplan, M.J. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 2011, 7, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.M.; Wilkinson, T.; McLoughlin, R.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.C.; Kim, N.D.; Sadik, C.D.; Seung, E.; Lan, Y.; Byrne, M.H.; Haribabu, B.; Iwakura, Y.; Luster, A.D. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 2010, 33, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Cao, W.; Yang, Y.; Liu, A. Extensive infiltration of neutrophils in the acute phase of experimental autoimmune encephalomyelitis in C57BL/6 mice. Histochem. Cell Biol. 2010, 133, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Demirci, S.; Kutluhan, S.; Koyuncuoglu, H.R.; Yurekli, V.A. The clinical significance of the neutrophil-to-lymphocyte ratio in multiple sclerosis. Int. J. Neurosci. 2015, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef]
- De Bondt, M.; Hellings, N.; Opdenakker, G.; Struyf, S. Neutrophils: Underestimated players in the pathogenesis of multiple sclerosis (MS). Int. J. Mol. Sci. 2020, 21, 4558. [Google Scholar] [CrossRef] [PubMed]
- Koiwa, M.; Goto, S.; Takahashi, K.; Kamada, T.; Takai, S.; Nakamura, H. Neutrophil/lymphocyte ratio in patients with rheumatoid arthritis treated with biological agents. J. Nippon. Med. Sch. 2016, 83, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Qin, B.; Hu, Z.; Ma, N.; Yang, M.; Wei, T.; Tang, Q.; Huang, Y.; Huang, F.; Liang, Y.; et al. Neutrophil-and platelet-to-lymphocyte ratios are correlated with disease activity in rheumatoid arthritis. Clin. Lab. 2015, 61, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Mercan, R.; Bitik, B.; Tufan, A.; Bozbulut, U.B.; Atas, N.; Ozturk, M.A.; Haznedaroglu, S.; Goker, B. The association between neutrophil/lymphocyte ratio and disease activity in rheumatoid arthritis and ankylosing spondylitis. J. Clin. Lab. Anal. 2016, 30, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yin, Y.; Kuai, S.; Shan, Z.; Pei, H.; Wang, J. Combination of neutrophil to lymphocyte ratio and platelet to lymphocyte ratio as diagnostic biomarker for rheumatoid arthritis. Int. J. Clin. Exp. Med. 2016, 9, 22076–22081. [Google Scholar]
- Auer, J.; Bläss, M.; Schulze-Koops, H.; Russwurm, S.; Nagel, T.; Kalden, J.R.; Röllinghoff, M.; Beuscher, H.U. Expression and regulation of CCL18 in synovial fluid neutrophils of patients with rheumatoid arthritis. Arthritis Res. Ther. 2007, 9, R94. [Google Scholar] [CrossRef] [Green Version]
- Cascão, R.; Rosário, H.; Souto-Carneiro, M.; Fonseca, J. Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun. Rev. 2010, 9, 531–535. [Google Scholar] [CrossRef]
- Tan, Y.; Qi, Q.; Lu, C.; Niu, X.; Bai, Y.; Jiang, C.; Wang, Y.; Zhou, Y.; Lu, A.; Xiao, C. Cytokine imbalance as a common mechanism in both psoriasis and rheumatoid arthritis. Mediat. Inflamm. 2017, 2017, 2405291. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes Type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [Green Version]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 455–463. [Google Scholar]
- Midgley, A.; McLaren, Z.; Moots, R.J.; Edwards, S.W.; Beresford, M.W. The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Wirestam, L.; Arve, S.; Linge, P.; Bengtsson, A.A. Neutrophils—Important communicators in systemic lupus erythematosus and antiphospholipid syndrome. Front. Immunol. 2019, 10, 2734. [Google Scholar] [CrossRef] [Green Version]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; MacKenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic targeting of the G-CSF receptor reduces neutrophil trafficking and joint inflammation in antibody-mediated inflammatory arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. IL-23-IL-17 immune axis: Discovery, mechanistic understanding, and clinical testing. Nat. Rev. Immunol. 2014, 14, 585. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- Robertson, J.D.; Ward, J.R.; Avila-Olias, M.; Battaglia, G.; Renshaw, S.A. Targeting neutrophilic inflammation using polymersome-mediated cellular delivery. J. Immunol. 2017, 198, 3596–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirivi, R.G.S.; Van Rosmalen, J.W.G.; Van Der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mócsai, A.; Zhou, M.; Meng, F.; Tybulewicz, V.; Lowell, C.A. Syk is required for integrin signaling in neutrophils. Immunity 2002, 16, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Volmering, S.; Block, H.; Boras, M.; Lowell, C.A.; Zarbock, A. The neutrophil btk signalosome regulates integrin activation during sterile inflammation. Immunity 2016, 44, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Okeke, E.B.; Louttit, C.; Fry, C.; Najafabadi, A.H.; Han, K.; Nemzek, J.; Moon, J.J. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials 2020, 238, 119836. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, L.; Karimi, N.; Morovati, S.; Alizadeh, K.; Kakish, J.E.; Vanderkamp, S.; Fazel, F.; Napoleoni, C.; Alizadeh, K.; Mehrani, Y.; et al. The Roles of Neutrophils in Cytokine Storms. Viruses 2021, 13, 2318. https://doi.org/10.3390/v13112318
Chan L, Karimi N, Morovati S, Alizadeh K, Kakish JE, Vanderkamp S, Fazel F, Napoleoni C, Alizadeh K, Mehrani Y, et al. The Roles of Neutrophils in Cytokine Storms. Viruses. 2021; 13(11):2318. https://doi.org/10.3390/v13112318
Chicago/Turabian StyleChan, Lily, Negar Karimi, Solmaz Morovati, Kasra Alizadeh, Julia E. Kakish, Sierra Vanderkamp, Fatemeh Fazel, Christina Napoleoni, Kimia Alizadeh, Yeganeh Mehrani, and et al. 2021. "The Roles of Neutrophils in Cytokine Storms" Viruses 13, no. 11: 2318. https://doi.org/10.3390/v13112318
APA StyleChan, L., Karimi, N., Morovati, S., Alizadeh, K., Kakish, J. E., Vanderkamp, S., Fazel, F., Napoleoni, C., Alizadeh, K., Mehrani, Y., Minott, J. A., Bridle, B. W., & Karimi, K. (2021). The Roles of Neutrophils in Cytokine Storms. Viruses, 13(11), 2318. https://doi.org/10.3390/v13112318