
Exploring the Diversity of the Human Blood Virome

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA/RNA Extraction and Amplification
2.3. Massive Parallel Sequencing
2.4. Sequence Analysis
2.5. Phylogenetic Analysis
2.6. Sanger Sequencing
2.7. Split Network Analysis
3. Results
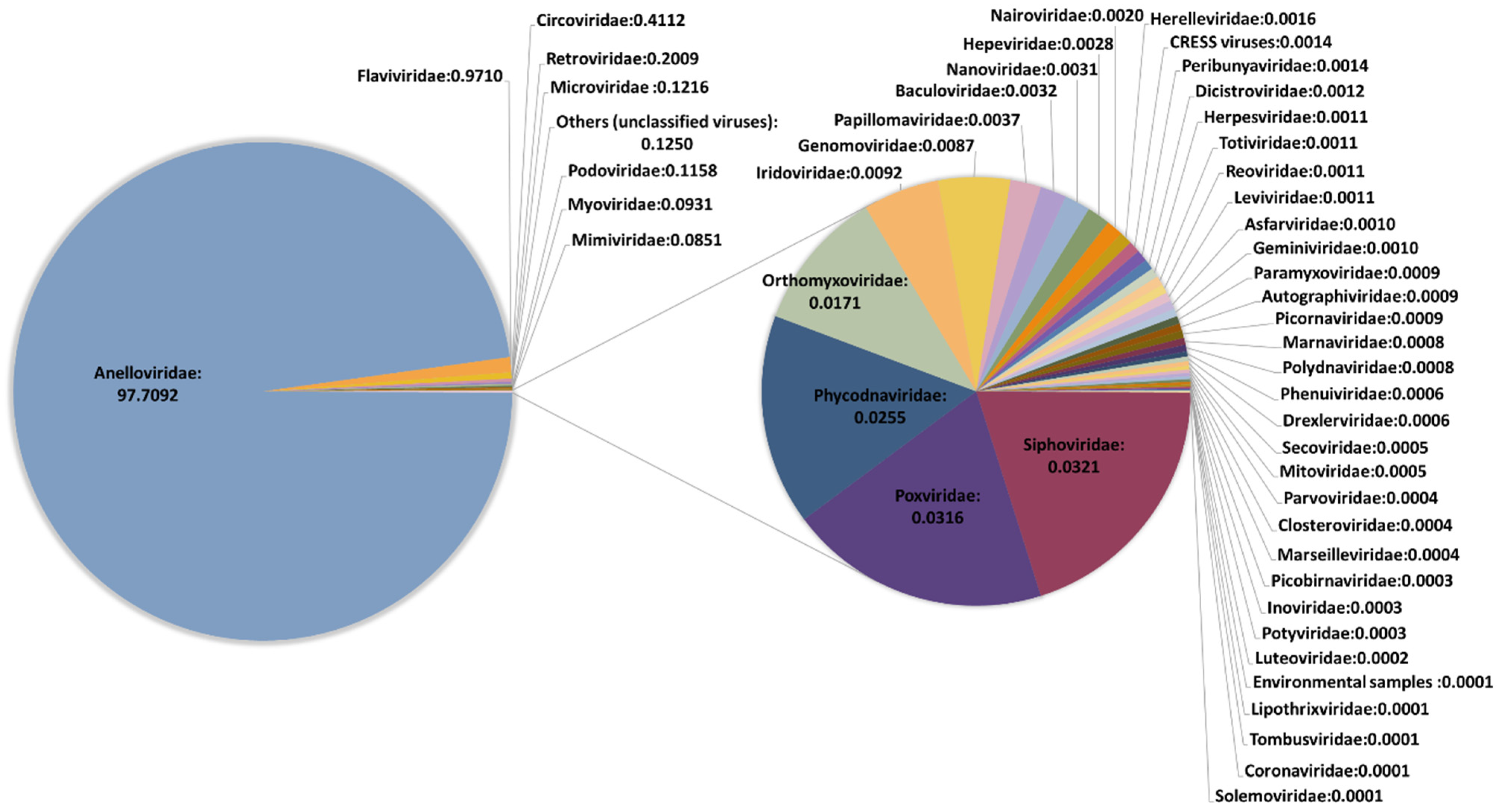
3.1. Overall Sequence Output
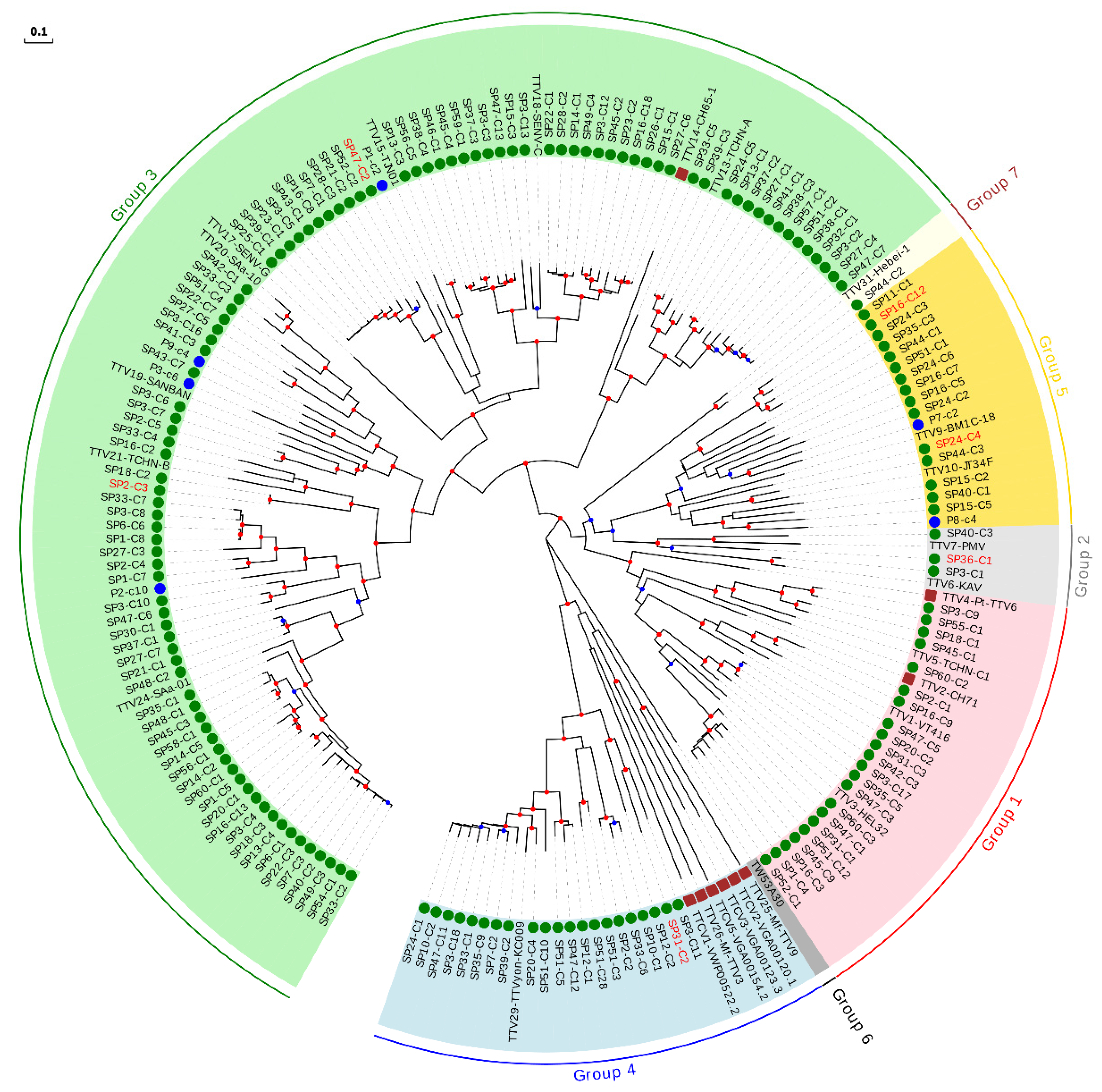
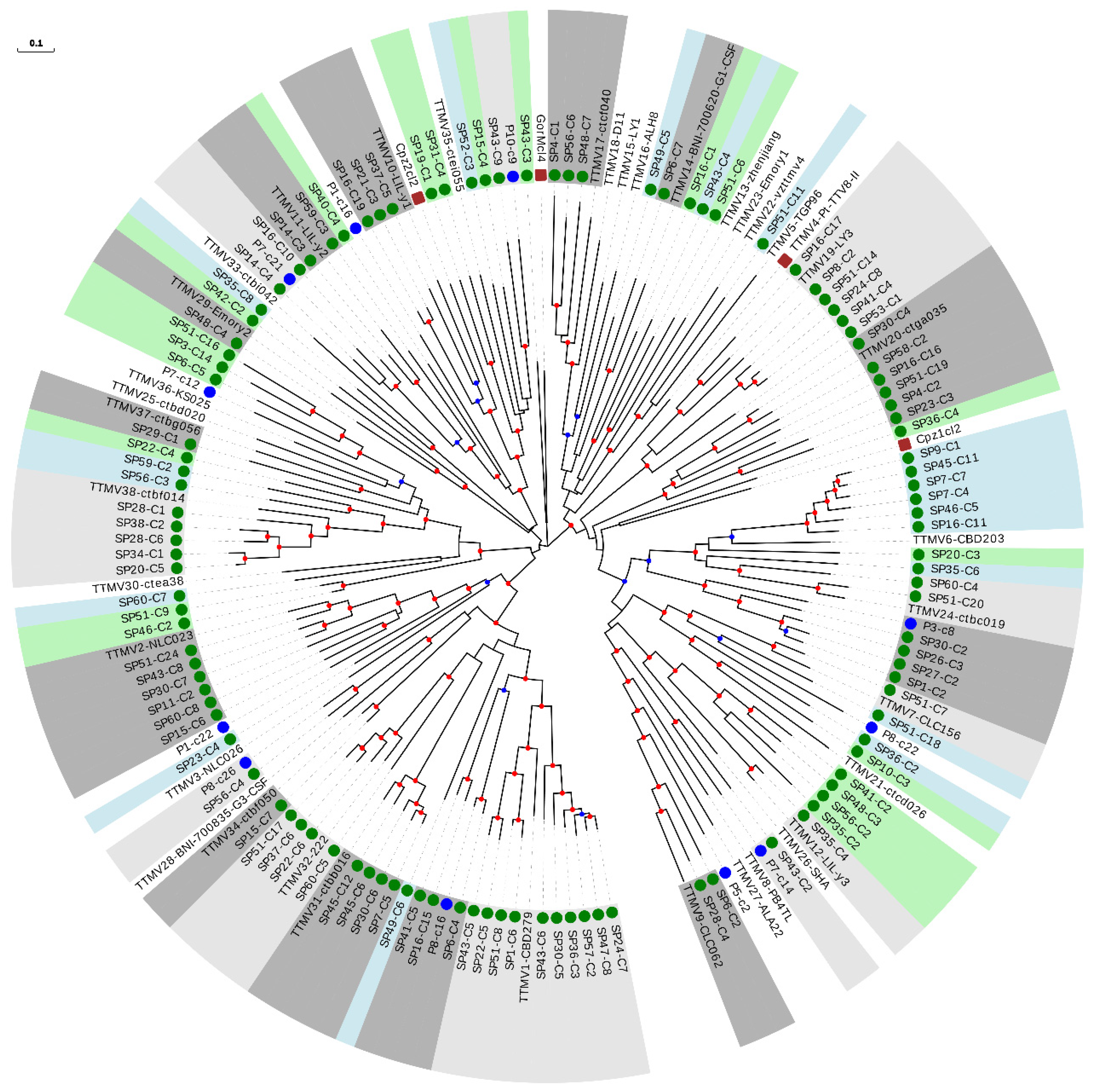
3.2. Phylogenetic Analysis of Anelloviruses
3.3. Analysis of HPgV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holmes, E.C. What does virus evolution tell us about virus origins? J. Virol. 2011, 85, 5247–5251. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Rambaut, A.; Andersen, K.G. Pandemics: Spend on surveillance, not prediction. Nature 2018, 558, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- French, R.K.; Holmes, E.C. An Ecosystems Perspective on Virus Evolution and Emergence. Trends Microbiol. 2020, 28, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479–480, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef]
- Furuta, R.A.; Sakamoto, H.; Kuroishi, A.; Yasiui, K.; Matsukura, H.; Hirayama, F. Metagenomic profiling of the viromes of plasma collected from blood donors with elevated serum alanine aminotransferase levels. Transfusion 2015, 55, 1889–1899. [Google Scholar] [CrossRef]
- Law, J.; Jovel, J.; Patterson, J.; Ford, G.; O’keefe, S.; Wang, W.; Meng, B.; Song, D.; Zhang, Y.; Tian, Z.; et al. Identification of Hepatotropic Viruses from Plasma Using Deep Sequencing: A Next Generation Diagnostic Tool. PLoS ONE 2013, 8, e60595. [Google Scholar] [CrossRef] [Green Version]
- Popgeorgiev, N.; Boyer, M.; Fancello, L.; Monteil, S.; Robert, C.; Rivet, R.; Nappez, C.; Azza, S.; Chiaroni, J.; Raoult, D.; et al. Marseillevirus-like virus recovered from blood donated by asymptomatic humans. J. Infect. Dis. 2013, 208, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.H.; Andersen, K.G.; Folarin, O.A.; Grove, J.N.; Odia, I.; Ehiane, P.E.; Omoniwa, O.; Omoregie, O.; Jiang, P.P.; Yozwiak, N.L.; et al. Discovery of Novel Rhabdoviruses in the Blood of Healthy Individuals from West Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003631. [Google Scholar] [CrossRef]
- Kapoor, A.; Kumar, A.; Simmonds, P.; Bhuva, N.; Chauhan, L.S.; Lee, B.; Sall, A.A.; Jin, Z.; Morse, S.S.; Shaz, B.; et al. Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with hepaciviruses and pegiviruses. MBio 2015, 6, e01466-15. [Google Scholar] [CrossRef] [Green Version]
- Spandole, S.; Berca, L.M.; Miha, G. Human anelloviruses: An update of molecular, epidemiological and clinical aspects. Arch. Virol. 2015, 160, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Stapleton, J.T. Tropism of human pegivirus (Formerly known as GB virus C/hepatitis G virus) and host immunomodulation: Insights into a highly successful viral infection. J. Gen. Virol. 2015, 96, 1521–1532. [Google Scholar] [CrossRef]
- Kaczorowska, J.; Hoek, L. Van Der. Human anelloviruses: Diverse, omnipresent and commensal members of the virome. FEMS Microbiol. Rev. 2020, 44, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, N.; Stapleton, J.T. GB virus C: The good boy virus? Trends Microbiol. 2012, 20, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Ataei, B.; Emami Naeini, A.; Khorvash, F.; Yazdani, M.R.; Javadi, A.-A. Prevalence of transfusion transmitted virus infection in hemodialysis patients and injection drug users compared to healthy blood donors in Isfahan, Iran. Gastroenterol. Res. Pract. 2012, 2012, 671927. [Google Scholar] [CrossRef] [Green Version]
- Cebriá-Mendoza, M.; Arbona, C.; Larrea, L.; Díaz, W.; Arnau, V.; Peña, C.; Bou, J.V.; Sanjuán, R.; Cuevas, J.M. Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans. Sci. Rep. 2021, 11, 6921. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining Chronic Viral Infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [Green Version]
- Tyschik, E.A.; Rasskazova, A.S.; Degtyareva, A.V.; Rebrikov, D.V.; Sukhikh, G.T. Torque teno virus dynamics during the first year of life. Virol. J. 2018, 15, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, E.L.; Stapleton, J.T. GB virus type C interactions with HIV: The role of envelope glycoproteins. J. Viral Hepat. 2009, 16, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhao, W.; Feng, Y.; Dai, J.; Li, Z.; Zhang, X.; Liu, L.; Bai, J.; Zhang, H.; Lu, L.; et al. A novel genotype of GB virus C: Its identification and predominance among injecting drug users in Yunnan, China. PLoS ONE 2011, 6, e21151. [Google Scholar] [CrossRef]
- Ghai, R.R.; Sibley, S.D.; Lauck, M.; Dinis, J.M.; Bailey, A.L.; Chapman, C.A.; Omeja, P.; Friedrich, T.C.; O’Connor, D.H.; Goldberg, T.L. Deep sequencing identifies two genotypes and high viral genetic diversity of human pegivirus (GB virus C) in rural Ugandan patients. J. Gen. Virol. 2013, 94, 2670–2678. [Google Scholar] [CrossRef] [Green Version]
- Lauck, M.; Bailey, A.L.; Andersen, K.G.; Goldberg, T.L.; Sabeti, P.C.; O’Connor, D.H. GB virus C coinfections in west African Ebola patients. J. Virol. 2015, 89, 2425–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, V.; Eloit, M. Viral metagenomics and blood safety. Transfus. Clin. Biol. 2016, 23, 28–38. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Martí, J.M. Recentrifuge: Robust comparative analysis and contamination removal for metagenomics. PLoS Comput. Biol. 2019, 15, e1006967. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Shoaib, M.; Baconnais, S.; Mechold, U.; Le Cam, E.; Lipinski, M.; Ogryzko, V. Multiple displacement amplification for complex mixtures of DNA fragments. BMC Genom. 2008, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Desnues, C.; Switzer, W.M.; Djoko, C.F.; Schneider, B.S.; Deng, X.; Delwart, E. Absence of giant blood Marseille-like virus DNA detection by polymerase chain reaction in plasma from healthy US blood donors and serum from multiply transfused patients from Cameroon. Transfusion 2015, 55, 1256–1262. [Google Scholar] [CrossRef]
- Sauvage, V.; Livartowski, A.; Boizeau, L.; Servant-Delmas, A.; Lionnet, F.; Lefrère, J.-J.; Laperche, S. No evidence of Marseillevirus-like virus presence in blood donors and recipients of multiple blood transfusions. J. Infect. Dis. 2014, 210, 2017–2018. [Google Scholar] [CrossRef] [Green Version]
- De Souza, W.M.; Fumagalli, M.J.; De Araujo, J.; Sabino-Santos, G., Jr.; Gonçalves, F.; Maia, M.; Farignoli, M.; Modha, S.; Schiavo, M.; Helena, L.; et al. Discovery of novel anelloviruses in small mammals expands the host range and diversity of the Anelloviridae. Virology 2018, 514, 9–17. [Google Scholar] [CrossRef]
- Ninomiya, M.; Nishizawa, T.; Takahashi, M.; Lorenzo, F.R.; Shimosegawa, T.; Okamoto, H. Identification and genomic characterization of a novel human torque teno virus of 3.2 kb. J. Gen. Virol. 2007, 88, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Wang, L.; Lin, C.; Liu, H. New Phylogenetic Groups of Torque Teno Virus Identified in Eastern Taiwan Indigenes. PLoS ONE 2016, 11, e0149901. [Google Scholar] [CrossRef] [PubMed]
- De Vlaminck, I.; Khush, K.K.; Strehl, C.; Kohli, B.; Luikart, H.; Neff, N.F.; Okamoto, J.; Snyder, T.M.; Cornfield, D.N.; Nicolls, M.R.; et al. Temporal Response of the Human Virome to Immunosuppression and Antiviral Therapy. Cell 2013, 155, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, M.; Takahashi, M.; Hoshino, Y.; Ichiyama, K.; Simmonds, P.; Okamoto, H. Analysis of the entire genomes of torque teno midi virus variants in chimpanzees: Infrequent cross-species infection between humans and chimpanzees. J. Gen. Virol. 2009, 90, 347–358. [Google Scholar] [CrossRef]
- Forns, X.; Fernández-Llama, P.; Costa, J.; López-Labrador, F.X.; Ampurdanés, S.; Olmedo, E.; Saiz, J.C.; Guilera, M.; López-Pedret, J.; Sánchez-Tapias, J.M.; et al. Hepatitis G virus infection in a haemodialysis unit: Prevalence and clinical implications. Nephrol. Dial. Transplant. 1997, 12, 956–960. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Zhang, Y.-Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for Novel Hepaciviruses in Rodents. PLOS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arze, C.A.; Springer, S.; Dudas, G.; Patel, S.; Bhattacharyya, A.; Swaminathan, H.; Brugnara, C.; Delagrave, S.; Ong, T.; Kahvejian, A.; et al. Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell Host Microbe 2021, 29, 11. [Google Scholar] [CrossRef] [PubMed]
- Thom, K.; Morrison, C.; Lewis, J.C.M.; Simmonds, P. Distribution of TT virus (TTV), TTV-like minivirus, and related viruses in humans and nonhuman primates. Virology 2003, 306, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Takahashi, M.; Nishizawa, T.; Tawara, A.; Fukai, K.; Muramatsu, U.; Naito, Y.; Yoshikawa, A. Genomic characterization of TT viruses (TTVs) in pigs, cats and dogs and their relatedness with species-specific TTVs in primates and tupaias. J. Gen. Virol. 2002, 83, 1291–1297. [Google Scholar] [CrossRef]
- Reshetnyak, V.I.; Maev, I.V.; Burmistrov, A.I.; Chekmazov, I.A.; Karlovich, T.I. Torque teno virus in liver diseases: On the way towards unity of view. World J. Gastroenterol. 2020, 26, 1691–1707. [Google Scholar] [CrossRef]
- Spandole-Dinu, S.; Cimponeriu, D.G.; Crăciun, A.-M.; Radu, I.; Nica, S.; Toma, M.; Alexiu, O.A.; Iorga, C.S.; Berca, L.; Nica, R. Prevalence of human anelloviruses in Romanian healthy subjects and patients with common pathologies. BMC Infect. Dis. 2018, 18, 334. [Google Scholar] [CrossRef]
- Strassl, R.; Schiemann, M.; Doberer, K.; Görzer, I.; Puchhammer-Stöckl, E.; Eskandary, F.; Kikić, Ž.; Gualdoni, G.A.; Vossen, M.G.; Rasoul-Rockenschaub, S.; et al. Quantification of Torque Teno Virus Viremia as a Prospective Biomarker for Infectious Disease in Kidney Allograft Recipients. J. Infect. Dis. 2018, 218, 1191–1199. [Google Scholar] [CrossRef]
- Frye, B.C.; Bierbaum, S.; Falcone, V.; Köhler, T.C.; Gasplmayr, M.; Hettich, I.; Dürk, T.; Idzko, M.; Zissel, G.; Hengel, H.; et al. Kinetics of Torque Teno Virus-DNA Plasma Load Predict Rejection in Lung Transplant Recipients. Transplantation 2019, 103, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Skewes-Cox, P.; Zhong, W.; Ganem, D.E.; Divers, T.J.; Van Blaricum, A.J.; Tennant, B.C.; Kistler, A.L. Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc. Natl. Acad. Sci. USA 2013, 110, E1407–E1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonsall, D.; Gregory, W.F.; Ip, C.L.C.; Donfield, S.; Iles, J.; Ansari, M.A.; Piazza, P.; Trebes, A.; Brown, A.; Frater, J.; et al. Evaluation of Viremia Frequencies of a Novel Human Pegivirus by Using Bioinformatic Screening and PCR. Emerg. Infect. Dis. 2016, 22, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A. Origin and evolution of GBV-C/hepatitis G virus and relationships with ancient human migrations. J. Mol. Evol. 2001, 53, 104–113. [Google Scholar] [CrossRef]
- Sharp, P.M.; Simmonds, P. Evaluating the evidence for virus/host co-evolution. Curr. Opin. Virol. 2011, 1, 436–441. [Google Scholar] [CrossRef]
- Marano, G.; Franchini, M.; Farina, B.; Piccinini, V.; Pupella, S.; Vaglio, S.; Grazzini, G.; Liumbruno, G.M. The human pegivirus: A new name for an “ancient” virus. Can transfusion medicine come up with something new? Acta Virol. 2017, 61, 401–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordier, F.; Deligny, M.-L.; Barré, R.; Robert, C.; Galicher, V.; Uch, R.; Fournier, P.-E.; Raoult, D.; Biagini, P. Human pegivirus isolates characterized by deep sequencing from hepatitis C virus-RNA and human immunodeficiency virus-RNA-positive blood donations, France. J. Med. Virol. 2018, 91, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parreira, R.; Branco, C.; Piedade, J.; Esteves, A. GB virus C (GBV-C) evolutionary patterns revealed by analyses of reference genomes, E2 and NS5B sequences amplified from viral strains circulating in the Lisbon area (Portugal). Infect. Genet. Evol. 2012, 12, 86–93. [Google Scholar] [CrossRef]
- Worobey, M.; Holmes, E.C. Homologous recombination in GB virus C/hepatitis G virus. Mol. Biol. Evol. 2001, 18, 254–261. [Google Scholar] [CrossRef]
- Blackard, J.T.; Ma, G.; Polen, C.; DuBois, J.C.; Gast, J.; Radens, C.M.; Sterling, R.K.; Sherman, K.E. Recombination among GB virus C (GBV-C) isolates in the United States. J. Gen. Virol. 2016, 97, 1537–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neibecker, M.; Schwarze-Zander, C.; Rockstroh, J.K.; Spengler, U.; Blackard, J.T. Evidence for extensive genotypic diversity and recombination of GB virus C (GBV-C) in Germany. J. Med. Virol. 2011, 83, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Padhi, A.; Xu, J.; Gong, X.; Tien, P. Evidence for within-host genetic recombination among the human pegiviral strains in HIV infected subjects. PLoS ONE 2016, 11, e0161880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Basaras, M.; Frost, S.; Haydon, D.; Cuceanu, N.; Prescott, L.; Kamenka, C.; Millband, D.; Sathar, M.A.; Simmonds, P. Phylogenetic analysis of GBV-C/hepatitis G virus. J. Gen. Virol. 2000, 81, 769–780. [Google Scholar] [CrossRef]
- Muerhoff, A.S.; Tillmann, H.L.; Manns, M.P.; Dawson, G.J.; Desai, S.M. GB virus C genotype determination in GB virus-C/HIV co-infected individuals. J. Med. Virol. 2003, 70, 141–149. [Google Scholar] [CrossRef]
- Schwarze-Zander, C.; Blackard, J.T.; Zheng, H.; Addo, M.M.; Lin, W.; Robbins, G.K.; Sherman, K.E.; Zdunek, D.; Hess, G.; Chung, R.T. GB virus C (GBV-C) infection in hepatitis C virus (HCV)/HIV-coinfected patients receiving HCV treatment: Importance of the GBV-C genotype. J. Infect. Dis. 2006, 194, 410–419. [Google Scholar] [CrossRef]
- Alcalde, R.; Nishiya, A.; Casseb, J.; Inocêncio, L.; Fonseca, L.A.M.; Duarte, A.J.S. Prevalence and distribution of the GBV-C/HGV among HIV-1-infected patients under anti-retroviral therapy. Virus Res. 2010, 151, 148–152. [Google Scholar] [CrossRef]
- Blackard, J.T.; Ma, G.; Welge, J.A.; Taylor, L.E.; Mayer, K.H.; Klein, R.S.; Celentano, D.D.; Sobel, J.D.; Jamieson, D.J.; King, C.C. Cytokine/chemokine expression associated with Human Pegivirus (HPgV) infection in women with HIV. J. Med. Virol. 2017, 89, 1904–1911. [Google Scholar] [CrossRef]
- Berzsenyi, M.D.; Bowden, D.S.; Roberts, S.K.; Revill, P.A. GB virus C genotype 2 predominance in a hepatitis C virus/HIV infected population associated with reduced liver disease. J. Gastroenterol. Hepatol. 2009, 24, 1407–1410. [Google Scholar] [CrossRef]
- Miao, Z.; Gao, L.; Song, Y.; Yang, M.; Zhang, M.; Lou, J.; Zhao, Y.; Wang, X.; Feng, Y.; Dong, X.; et al. Prevalence and Clinical Impact of Human Pegivirus-1 Infection in HIV-1-Infected Individuals in Yunnan, China. Viruses 2017, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, S.; Schmidt, R.; Day, T. Fighting the Public Health Burden of AIDS With the Human Pegivirus. Am. J. Epidemiol. 2019, 188, 1586–1594. [Google Scholar] [CrossRef]
- Maidana-Giret, M.T.; Silva, T.M.; Sauer, M.M.; Tomiyama, H.; Levi, J.E.; Bassichetto, K.C.; Nishiya, A.; Diaz, R.S.; Sabino, E.C.; Palacios, R.; et al. GB virus type C infection modulates T-cell activation independently of HIV-1 viral load. AIDS 2009, 23, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Rydze, R.T.; Chivero, E.T.; Stapleton, J.T. GB virus C viremia is associated with higher levels of double-negative T cells and lower T-cell activation in HIV-infected individuals receiving antiretroviral therapy. J. Infect. Dis. 2012, 206, 1469–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.M.; Stapleton, J.T.; Klinzman, D.; McLinden, J.H.; Purdue, M.P.; Katki, H.A.; Engels, E.A. GBV-C infection and risk of NHL among U.S. adults. Cancer Res. 2014, 74, 5553–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajden, M.; Yu, A.; Braybrook, H.; Lai, A.S.; Mak, A.; Chow, R.; Cook, D.; Tellier, R.; Petric, M.; Gascoyne, R.D.; et al. GBV-C/hepatitis G virus infection and non-Hodgkin lymphoma: A case control study. Int. J. Cancer 2010, 126, 2885–2892. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.H.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Sanchez Leon, M.; Jain, K.; et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. MBio 2013, 4, e00216-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibley, S.D.; Lauck, M.; Bailey, A.L.; Hyeroba, D.; Tumukunde, A.; Weny, G.; Chapman, C.A.; O’Connor, D.H.; Goldberg, T.L.; Friedrich, T.C. Discovery and characterization of distinct simian pegiviruses in three wild African Old World monkey species. PLoS ONE 2014, 9, e98569. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool | Anellovirus Reads | Pegivirus Reads | Other Viruses | Pool/Blank | Anellovirus Reads | Pegivirus Reads | Other Viruses |
|---|---|---|---|---|---|---|---|
| SP1 | 101,069 | 25,965 | 64 | SP35 | 419,986 | 0 | 317 |
| SP2 | 1,580,534 | 0 | 3013 | SP36 | 185,281 | 0 | 144 |
| SP3 | 131,969 | 3669 | 421 | SP37 | 666,063 | 0 | 1311 |
| SP4 | 9992 | 4250 | 61 | SP38 | 242,853 | 2261 | 479 |
| SP5 | 47,927 | 0 | 225 | SP39 | 15,756 | 0 | 200 |
| SP6 | 718,633 | 0 | 330 | SP40 | 342,193 | 0 | 3390 |
| SP7 | 63,139 | 0 | 80 | SP41 | 169,614 | 0 | 2815 |
| SP8 | 76,204 | 0 | 5089 | SP42 | 4519 | 0 | 118 |
| SP9 | 153,491 | 0 | 52 | SP43 | 206,185 | 0 | 99 |
| SP10 | 30,175 | 0 | 1649 | SP44 | 7975 | 10,713 | 19 |
| SP11 | 9787 | 5706 | 143 | SP45 | 124,171 | 0 | 210 |
| SP12 | 57,559 | 0 | 15,397 | SP46 | 29,728 | 0 | 431 |
| SP13 | 95,922 | 1173 | 4844 | SP47 | 150,531 | 0 | 3731 |
| SP14 | 271,731 | 0 | 1757 | SP48 | 45,430 | 0 | 676 |
| SP15 | 141,896 | 0 | 37 | SP49 | 94,919 | 5226 | 255 |
| SP16 | 149,985 | 9 | 2610 | SP50 | 0 | 0 | 340 |
| SP17 | 10 | 0 | 74 | SP51 | 299,530 | 0 | 17 |
| SP18 | 24,134 | 0 | 7168 | SP52 | 59,852 | 0 | 16 |
| SP19 | 74,391 | 339 | 9506 | SP53 | 14,323 | 5344 | 68 |
| SP20 | 73,067 | 373 | 21 | SP54 | 2404 | 0 | 7634 |
| SP21 | 124,389 | 0 | 4952 | SP55 | 663 | 0 | 121 |
| SP22 | 51,168 | 0 | 3428 | SP56 | 25,673 | 3523 | 131 |
| SP23 | 51,730 | 0 | 557 | SP57 | 52,296 | 0 | 1737 |
| SP24 | 71,389 | 0 | 7583 | SP58 | 1232 | 2158 | 241 |
| SP25 | 4269 | 0 | 262 | SP59 | 157,753 | 0 | 46 |
| SP26 | 27,676 | 0 | 84 | SP60 | 36,470 | 0 | 324 |
| SP27 | 7659 | 0 | 3030 | C01 | 0 | 0 | 593 |
| SP28 | 96,187 | 0 | 270 | C02 | 0 | 0 | 8022 |
| SP29 | 334,689 | 6606 | 18,366 | C03 | 0 | 0 | 76,410 |
| SP30 | 69,110 | 6924 | 156 | C04 | 0 | 0 | 3589 |
| SP31 | 332,437 | 0 | 816 | C05 | 0 | 0 | 93,531 |
| SP32 | 1011 | 0 | 223 | C06 | 0 | 0 | 4588 |
| SP33 | 72,784 | 2033 | 68 | C07 | 0 | 0 | 2964 |
| SP34 | 270,083 | 0 | 57 | C09 | 0 | 0 | 6731 |
| Cebriá et al. (2021) 2 | This Study 3 | ||||||
|---|---|---|---|---|---|---|---|
| Species 1 | Sequences 4 | Novel Species 5 | Coincident Clusters (%) 6 | Sequences 4 | Novel Species 5 | Coincident Clusters (%) 6 | |
| TTV | 26 | 68 | 6 (8.8) | 13 (50.0) | 160 | 6 (3.8) | 20 (62.5) |
| TTMV | 38 | 29 | 11 (37.9) | 11 (28.9) | 111 | 27 (24.3) | 24 (49.0) |
| TTMDV | 15 | 17 | 9 (52.9) | 5 (33.3) | 61 | 17 (27.9) | 16 (66.6) |
| Total | 79 | 114 | 26 (22.8) | 29 (36.7) | 332 | 50 (15.1) | 60 (57.1) |
| Sample/Pool | # Reads | Average Depth Coverage | Genome Coverage | Polyprotein Coverage |
|---|---|---|---|---|
| SP1 | 25,965 | 1010.7 | 98.4 | 98.6 |
| SP3 | 3669 | 130.1 | 94.2 | 95.6 |
| SP4 | 4250 | 157.7 | 92.5 | 93.8 |
| SP11 | 5706 | 204.0 | 99.2 | 100.0 |
| SP13 | 1173 | 40.7 | 96.4 | 97.4 |
| SP16 | 9 | 2.4 | 5.5 | 6.0 |
| SP19 | 339 | 12.4 | 82.7 | 89.9 |
| SP20 | 373 | 13.5 | 70.2 | 76.4 |
| SP29 | 6606 | 228.7 | 98.2 | 99.2 |
| SP30 | 6924 | 230.1 | 89.7 | 91.1 |
| SP33 | 2033 | 71.1 | 92.6 | 94.8 |
| SP38 | 2261 | 82.7 | 99.6 | 100.0 |
| SP44 | 10,713 | 392.0 | 99.1 | 100.0 |
| SP49 | 5226 | 165.7 | 83.4 | 84.0 |
| SP53 * | 5344 | 181.2 | 97.5 | 99.8 |
| SP56 | 3523 | 121.1 | 91.7 | 93.3 |
| SP58 | 2158 | 73.2 | 93.6 | 94.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cebriá-Mendoza, M.; Bracho, M.A.; Arbona, C.; Larrea, L.; Díaz, W.; Sanjuán, R.; Cuevas, J.M. Exploring the Diversity of the Human Blood Virome. Viruses 2021, 13, 2322. https://doi.org/10.3390/v13112322
Cebriá-Mendoza M, Bracho MA, Arbona C, Larrea L, Díaz W, Sanjuán R, Cuevas JM. Exploring the Diversity of the Human Blood Virome. Viruses. 2021; 13(11):2322. https://doi.org/10.3390/v13112322
Chicago/Turabian StyleCebriá-Mendoza, María, María A. Bracho, Cristina Arbona, Luís Larrea, Wladimiro Díaz, Rafael Sanjuán, and José M. Cuevas. 2021. "Exploring the Diversity of the Human Blood Virome" Viruses 13, no. 11: 2322. https://doi.org/10.3390/v13112322
APA StyleCebriá-Mendoza, M., Bracho, M. A., Arbona, C., Larrea, L., Díaz, W., Sanjuán, R., & Cuevas, J. M. (2021). Exploring the Diversity of the Human Blood Virome. Viruses, 13(11), 2322. https://doi.org/10.3390/v13112322