
Mass Spectrometry-Based System for Identifying and Typing Norovirus Major Capsid Protein VP1

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Norovirus VP1-cDNA Preparation
2.2. The VP1 Protein Expression and Purification
2.3. Western Blotting for VP1 Protein
2.4. Electron Microscopy for Morphological Study of VP1 Self-Assembled VLP
2.5. Matrix-Assisted Laser Desorption Ionization – Time of Flight (MALDI-TOP) MS for Examining Intact VP1 Protein
2.6. Ultra-Performance Liquid Chromatography (UPLC/MSE) Analysis of Peptide Sequences of Digested-VP1 Protein
2.7. UPLC/MSE Data Analysis
2.8. Likelihood-Mapping Analysis
2.9. Coevolution Analysis
3. Results
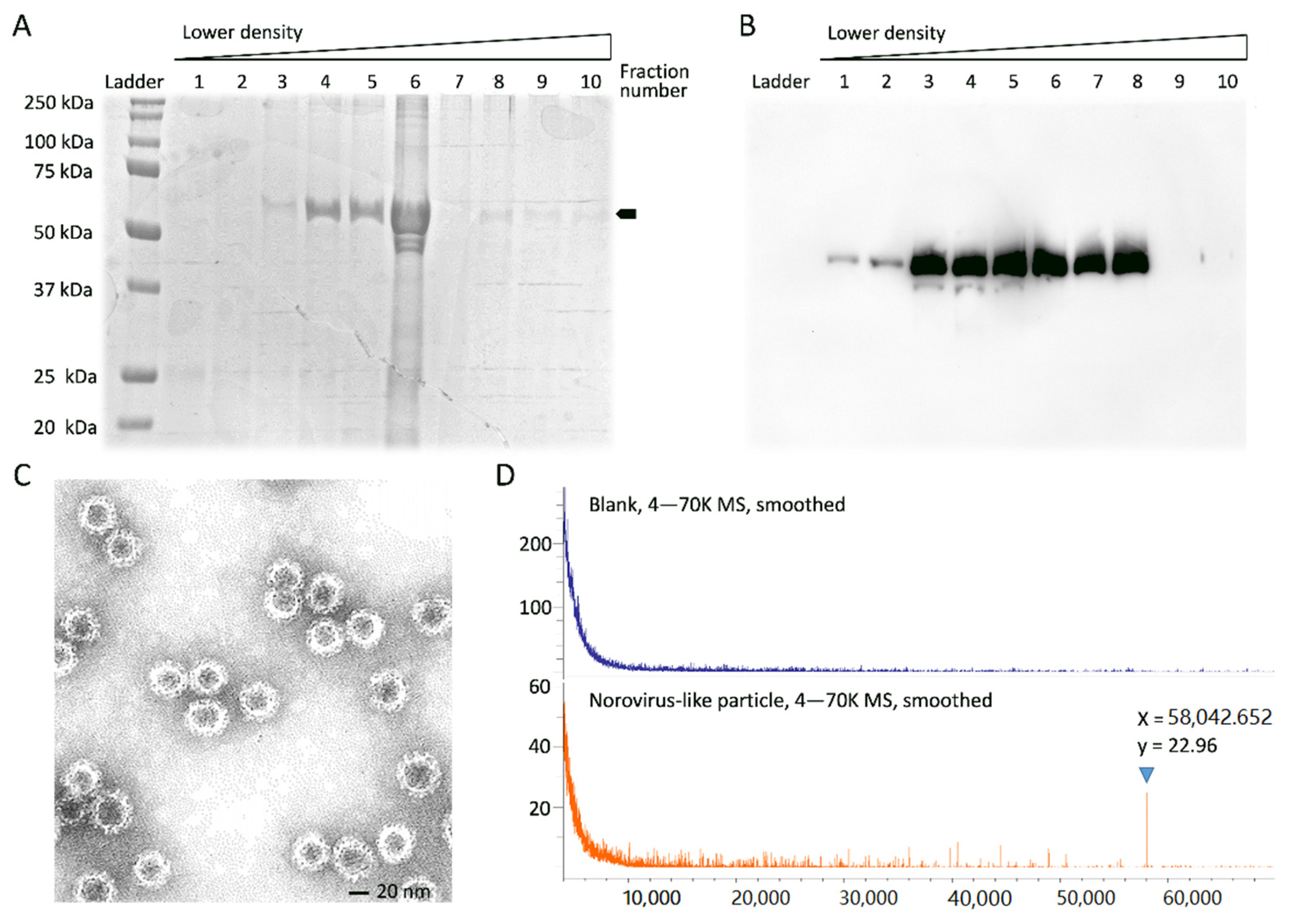
3.1. Expression and Characterization of Norovirus VP1 Protein
3.2. VP1 Peptide Sequencing by LC/MSE
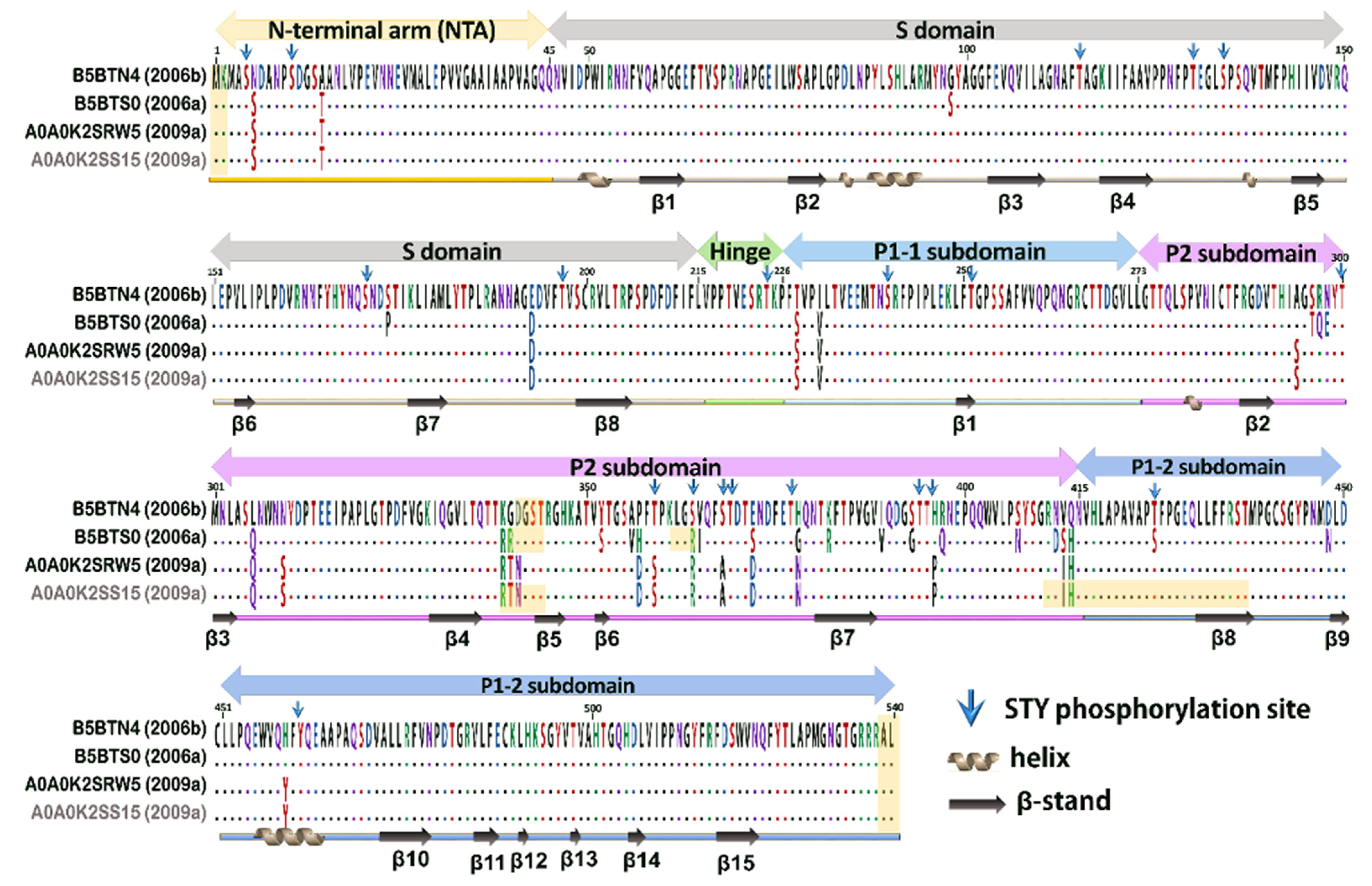
3.3. Post-Translational Modification Analysis
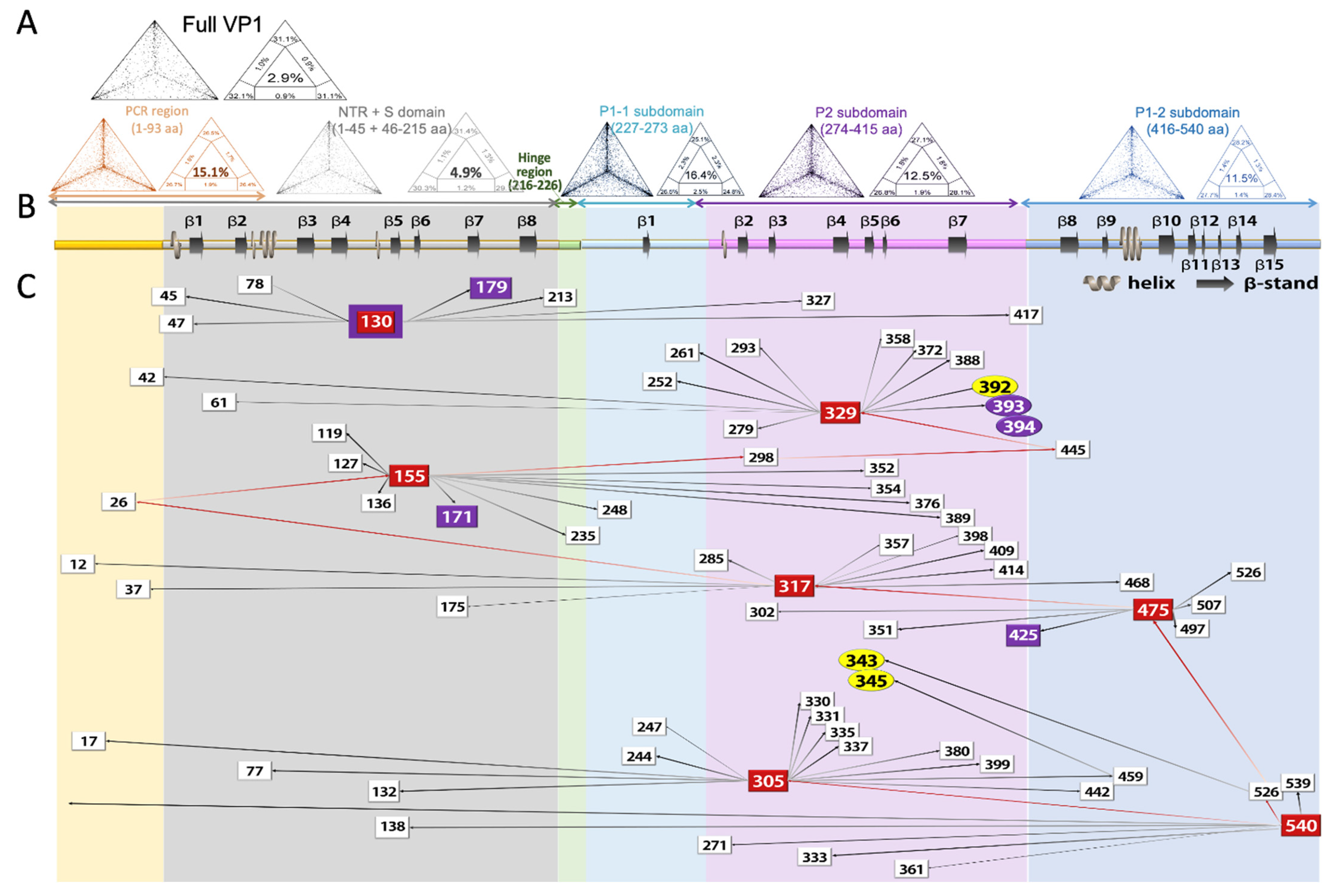
3.4. Likelihood-Mapping Analysis
3.5. Co-Evalution Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glass, R.I.; Parashar, U.D.; Estes, M.K. Norovirus gastroenteritis. N. Engl. J. Med. 2009, 361, 1776–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Graaf, M.; van Beek, J.; Koopmans, M.P. Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 2016, 14, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Bonifacio, J.; Bucardo, F.; Buesa, J.; Bruggink, L.; Chan, M.C.-W.; Fumian, T.M.; Giri, S.; Gonzalez, M.D.; Hewitt, J.; et al. Global Trends in Norovirus Genotype Distribution among Children with Acute Gastroenteritis. Emerg. Infect. Dis. 2021, 27, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Hall, A.J.; Vinje, J.; Parashar, U.D. Noroviruses: A comprehensive review. J. Clin. Virol. 2009, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.; et al. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti-Ciarlet, A.; White, L.J.; Chen, R.; Prasad, B.V.V.; Estes, M.K. Structural Requirements for the Assembly of Norwalk Virus-Like Particles. J. Virol. 2002, 76, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.-P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Rockx, B.H.G.; Vennema, H.; Hoebe, C.; Duizer, E.; Koopmans, M.P.G. Association of Histo–Blood Group Antigens and Susceptibility to Norovirus Infections. J. Infect. Dis. 2005, 191, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Xia, M.; Chen, Y.; Bu, W.; Hegde, R.S.; Meller, J.; Li, X.; Jiang, X. Conservation of Carbohydrate Binding Interfaces—Evidence of Human HBGA Selection in Norovirus Evolution. PLoS ONE 2009, 4, e5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeini, H.; Afridi, S.Q.; Donakonda, S.; Knolle, P.A.; Protzer, U.; Hoffmann, D. Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies. Vaccines 2021, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Hutson, A.M.; Estes, M.K.; Prasad, B.V. Atomic resolution structural characterization of recognition of his-to-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. USA 2008, 105, 9175–9180. [Google Scholar] [CrossRef] [Green Version]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus Immunity and the Great Escape. PLoS Pathog. 2012, 8, e1002921. [Google Scholar] [CrossRef] [Green Version]
- Straub, T.M.; Zu Bentrup, K.H.; Coghlan, P.O.; Dohnalkova, A.; Mayer, B.K.; Bartholomew, R.A.; Valdez, C.O.; Bruckner-Lea, C.J.; Gerba, C.P.; Abbaszadegan, M.A.; et al. In Vitro Cell Culture Infectivity Assay for Human Noroviruses. Emerg. Infect. Dis. 2007, 13, 396–403. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Baron, E.J.; Miller, J.M.; Weinstein, M.P.; Richter, S.S.; Gilligan, P.H.; Thomson, R.B., Jr.; Bourbeau, P.; Carroll, K.C.; Kehl, S.C.; Dunne, W.M.; et al. A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2013 recommendations by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM)(a). Clin. Infect. Dis. 2013, 57, e22–e121. [Google Scholar] [CrossRef] [Green Version]
- Vinjé, J. Advances in Laboratory Methods for Detection and Typing of Norovirus. J. Clin. Microbiol. 2014, 53, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Conti, B.J.; Leicht, A.S.; Kirchdoerfer, R.N.; Sussman, M.R. Mass spectrometric based detection of protein nucleotidylation in the RNA polymerase of SARS-CoV-2. Commun. Chem. 2021, 4, 41. [Google Scholar] [CrossRef]
- Chen, B.; Xiao, G.; He, M.; Hu, B. Elemental Mass Spectrometry and Fluorescence Dual-Mode Strategy for Ultrasensitive Label-Free Detection of HBV DNA. Anal. Chem. 2021, 93, 9454–9461. [Google Scholar] [CrossRef] [PubMed]
- Oyama, H.; Ishii, K.; Maruno, T.; Torisu, T.; Uchiyama, S. Characterization of adeno-associated virus capsid proteins with two types of VP3 related components by capillary gel electrophoresis and mass spectrometry. Hum. Gene Ther. 2021, 32, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Mallagaray, A.; Creutznacher, R.; Dülfer, J.; Mayer, P.H.O.; Grimm, L.L.; Orduña, J.M.; Trabjerg, E.; Stehle, T.; Rand, K.D.; Blaum, B.S.; et al. A post-translational modification of human Norovirus capsid protein attenuates glycan binding. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavan, S.; Mangalaparthi, K.K.; Singh, S.; Renuse, S.; Vanderboom, P.M.; Madugundu, A.K.; Budhraja, R.; McAulay, K.; Grys, T.E.; Rule, A.D.; et al. Mass Spectrometric Analysis of Urine from COVID-19 Patients for Detection of SARS-CoV-2 Viral Antigen and to Study Host Response. J. Proteome Res. 2021, 20, 3404–3413. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Lindberg, J.; Shevchenko, G.; Gustavsson, I.; Bergquist, J.; Gyllensten, U.; Enroth, S. Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS. Cancers 2021, 13, 2592. [Google Scholar] [CrossRef] [PubMed]
- Motomura, K.; Yokoyama, M.; Ode, H.; Nakamura, H.; Mori, H.; Kanda, T.; Oka, T.; Katayama, K.; Noda, M.; Tanaka, T.; et al. Divergent evolution of norovirus GII/4 by genome recombination from May 2006 to February 2009 in Japan. J. Virol. 2010, 84, 8085–8097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, U.; Kuharev, J.; Navarro, P.; Levin, Y.; Schild, H.; Tenzer, S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2013, 11, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, K.; Goshe, M.B. Challenges and strategies for targeted phosphorylation site identification and quantification using mass spectrometry analysis. Briefings Funct. Genom. Proteom. 2009, 8, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; von Haeseler, A. Maximum-Likelihood Analysis Using TREE-PUZZLE. Current protocols in bioinformatics 2007, Chapter 6, Unit 6.6. Available online: https://currentprotocols.onlineli-brary.wiley.com/doi/abs/10.1002/0471250953.bi0606s17 (accessed on 18 November 2021).
- Poon, A.F.Y.; Lewis, F.I.; Frost, S.D.W.; Pond, S.L.K. Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef] [Green Version]
- Strimmer, K.; von Haeseler, A. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Colquhoun, D.; Schwab, K.J.; Cole, R.N.; Halden, R.U. Detection of Norovirus Capsid Protein in Authentic Standards and in Stool Extracts by Matrix-Assisted Laser Desorption Ionization and Nanospray Mass Spectrometry. Appl. Environ. Microbiol. 2006, 72, 2749–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Fu, M.; Hu, Q. Advances in Human Norovirus Vaccine Research. Vaccines 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Kao, C.C. Non-encapsidation activities of the capsid proteins of positive-strand RNA viruses. Virology 2013, 446, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Turiño, S.; Pérez, J.D.J.; Hervás, M.; Navajas, R.; Ciordia, S.; Udeshi, N.D.; Shabanowitz, J.; Hunt, D.F.; García, J.A. Phosphorylation coexists with O-GlcNAcylation in a plant virus protein and influences viral infection. Mol. Plant Pathol. 2018, 19, 1427–1443. [Google Scholar] [CrossRef] [Green Version]
- Vongpunsawad, S.; Prasad, B.; Estes, M.K. Norwalk Virus Minor Capsid Protein VP2 Associates within the VP1 Shell Domain. J. Virol. 2013, 87, 4818–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfraction Number | Refractive Index | CsCl Density (g/cm3) |
|---|---|---|
| 1 | 1.3626 | 1.3006 |
| 2 | 1.3633 | 1.3082 |
| 3 | 1.3639 | 1.3147 |
| 4 | 1.3648 | 1.3245 |
| 5 | 1.3653 | 1.3299 |
| 6 | 1.3658 | 1.3353 |
| 7 | 1.3666 | 1.3440 |
| 8 | 1.3667 | 1.3451 |
| 9 | 1.3679 | 1.3581 |
| 10 | 1.3689 | 1.3690 |
| H2O | 1.3325 | 0.9737 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, P.-Y.; Huang, H.-W.; Boonchan, M.; Tyan, Y.-C.; Louis, K.L.; Lee, K.-M.; Motomura, K.; Ke, L.-Y. Mass Spectrometry-Based System for Identifying and Typing Norovirus Major Capsid Protein VP1. Viruses 2021, 13, 2332. https://doi.org/10.3390/v13112332
Chu P-Y, Huang H-W, Boonchan M, Tyan Y-C, Louis KL, Lee K-M, Motomura K, Ke L-Y. Mass Spectrometry-Based System for Identifying and Typing Norovirus Major Capsid Protein VP1. Viruses. 2021; 13(11):2332. https://doi.org/10.3390/v13112332
Chicago/Turabian StyleChu, Pei-Yu, Hui-Wen Huang, Michittra Boonchan, Yu-Chang Tyan, Kevin Leroy Louis, Kun-Mu Lee, Kazushi Motomura, and Liang-Yin Ke. 2021. "Mass Spectrometry-Based System for Identifying and Typing Norovirus Major Capsid Protein VP1" Viruses 13, no. 11: 2332. https://doi.org/10.3390/v13112332
APA StyleChu, P. -Y., Huang, H. -W., Boonchan, M., Tyan, Y. -C., Louis, K. L., Lee, K. -M., Motomura, K., & Ke, L. -Y. (2021). Mass Spectrometry-Based System for Identifying and Typing Norovirus Major Capsid Protein VP1. Viruses, 13(11), 2332. https://doi.org/10.3390/v13112332