
Presence and Persistence of Putative Lytic and Temperate Bacteriophages in Vaginal Metagenomes from South African Adolescents

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Sample Selection for This Sub-Study
2.3. DNA Sample Preparation and Whole Community Metagenomic Sequencing
2.4. Sequence Processing and Annotation
2.5. In Silico Identification of Virulent Bacteriophages in Vaginal Metagenomes
2.6. In Silico Identification and Characterisation of Prophages in Vaginal Metagenomes
2.7. Identification of CRISPR Repeats/Spacers, Predicted Cas Proteins, and CRISPR-Cas Systems
2.8. Data Analysis
3. Results
3.1. Cohort Characteristics
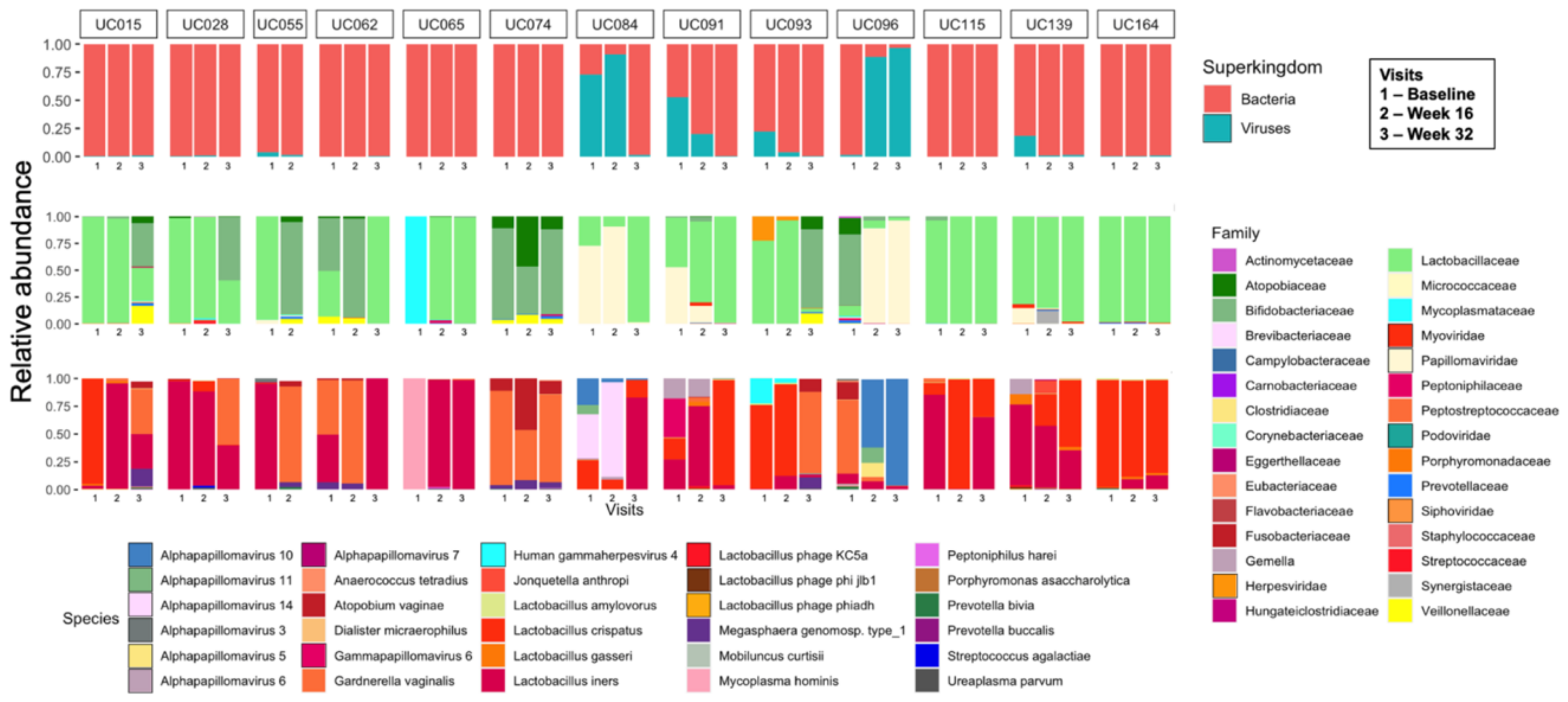
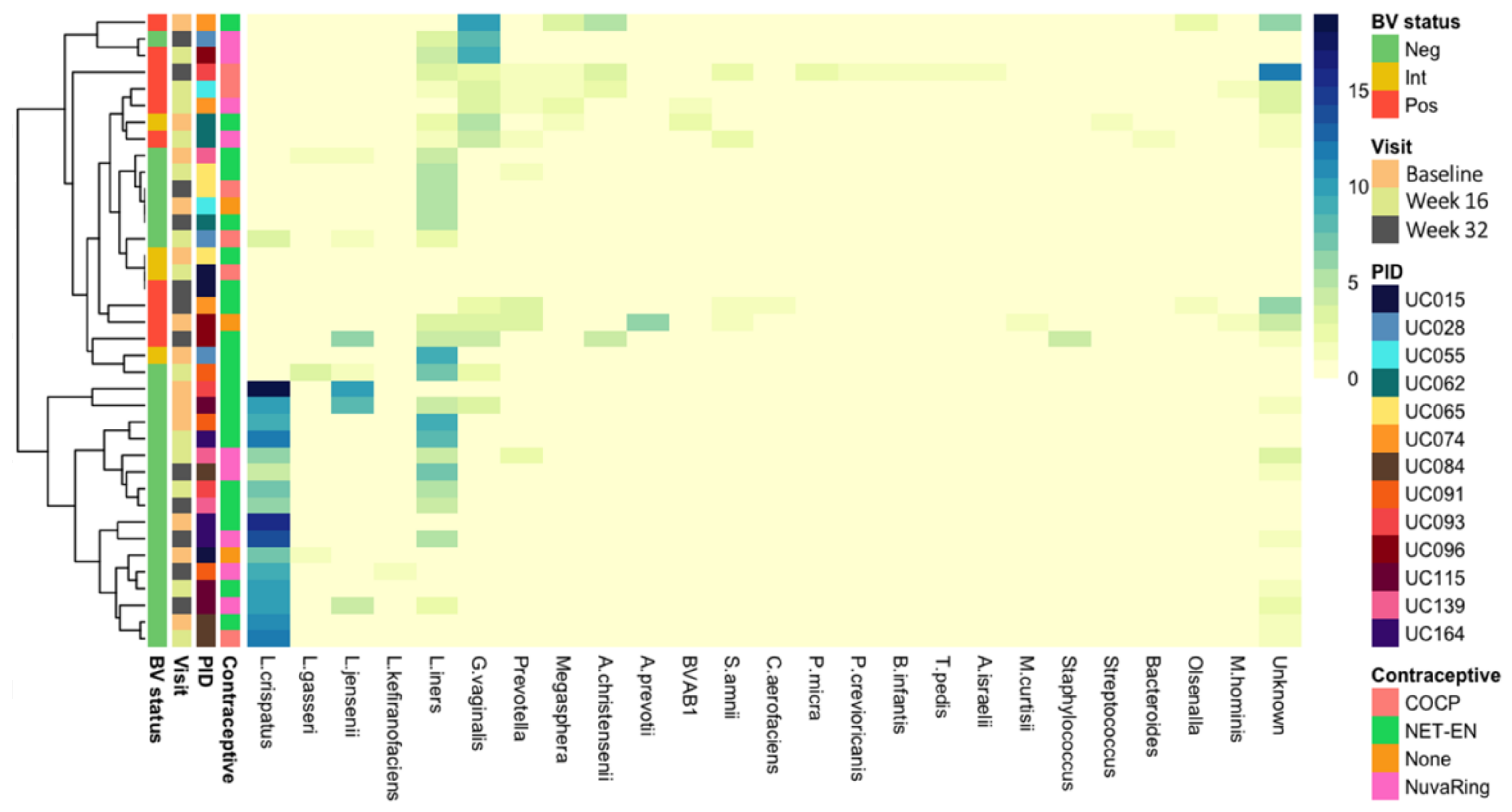
3.2. Vaginal Metagenomes of South African Adolescents
3.3. Identification of Prokaryote-Infecting Viruses in the Metagenomes of South African Adolescents
3.4. Presence of CRISPR Loci
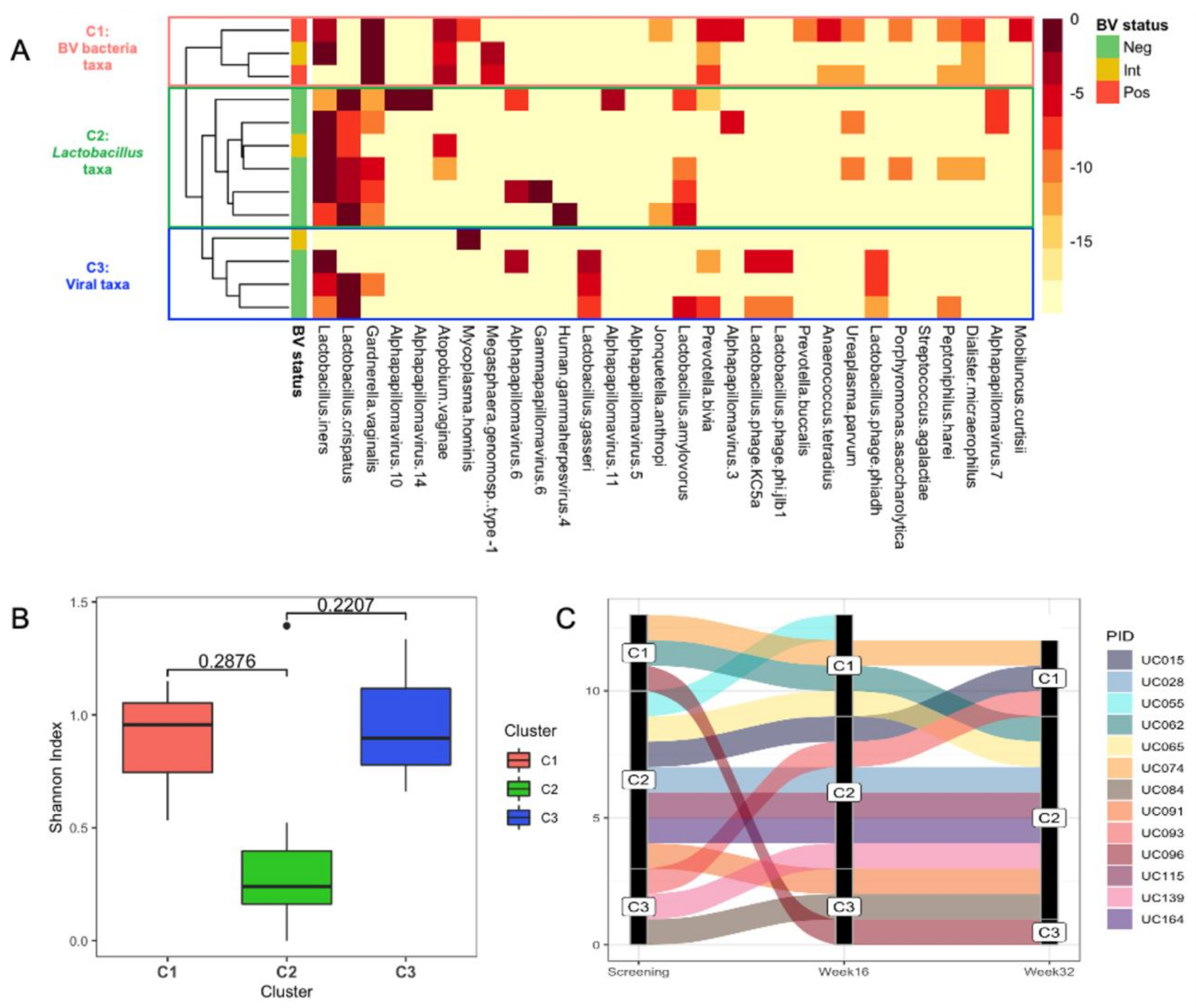
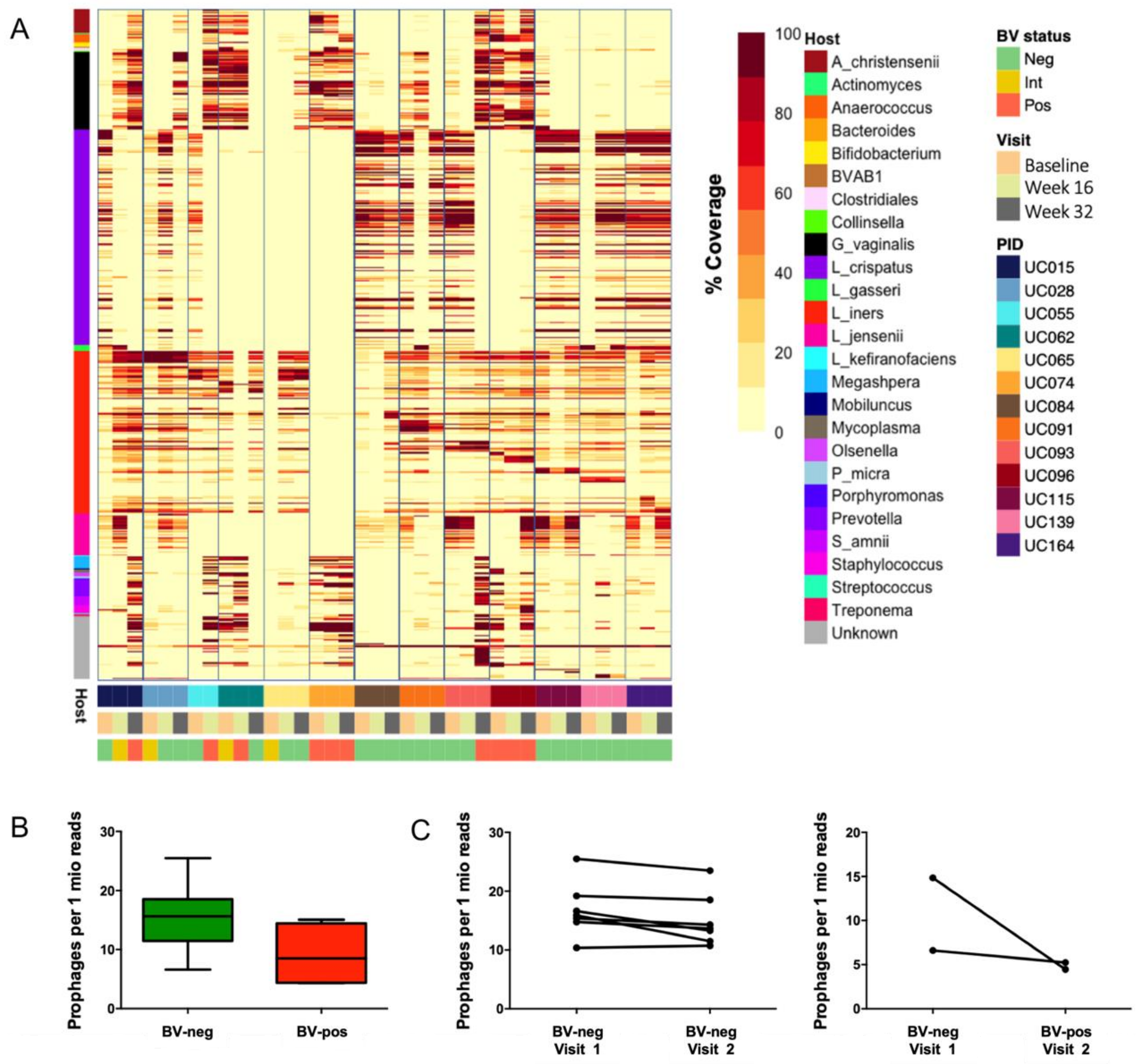
3.5. Identification of Putative Prophages in the Metagenomes of South African Adolescents and Associations with Vaginal Microbiota Stability
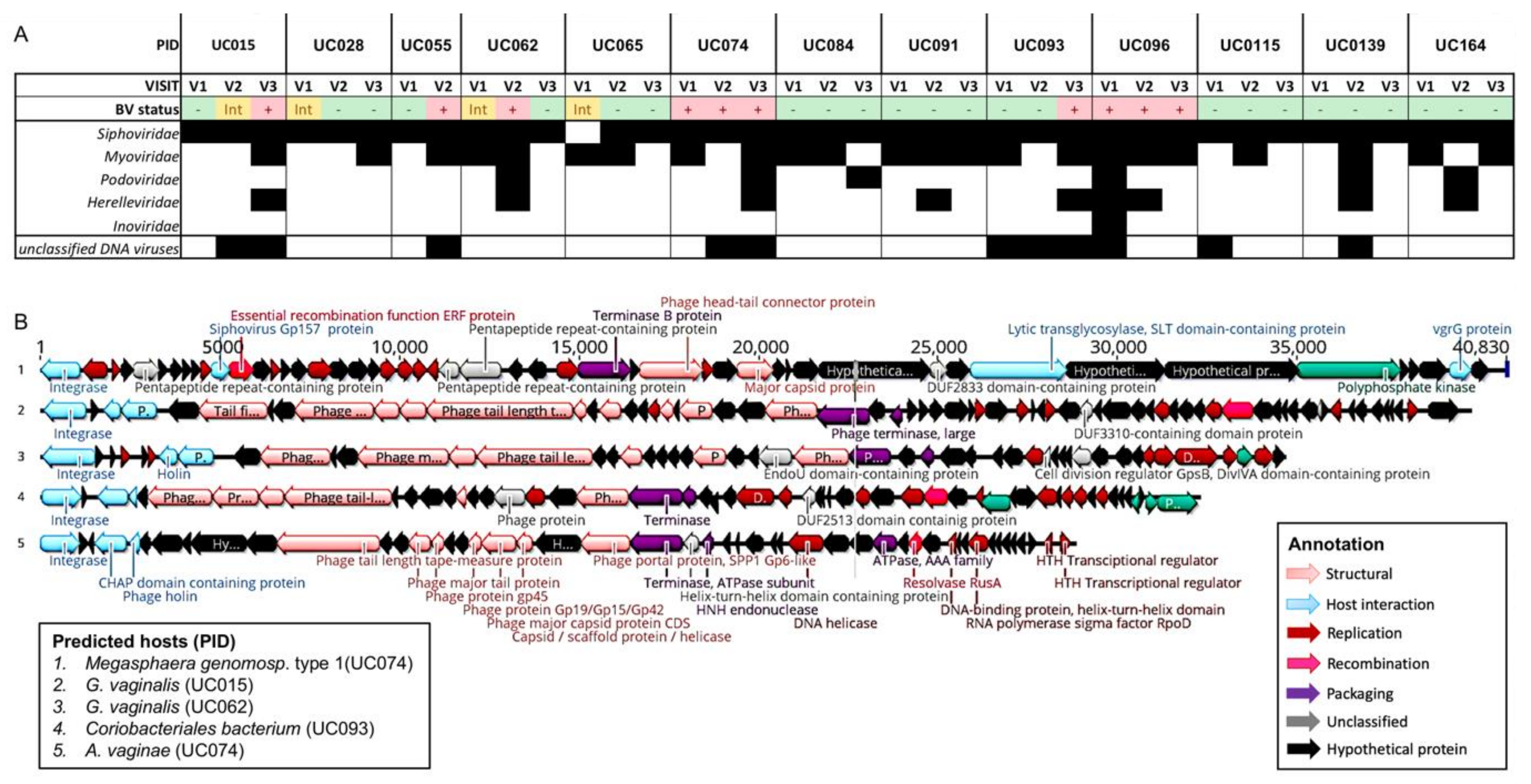
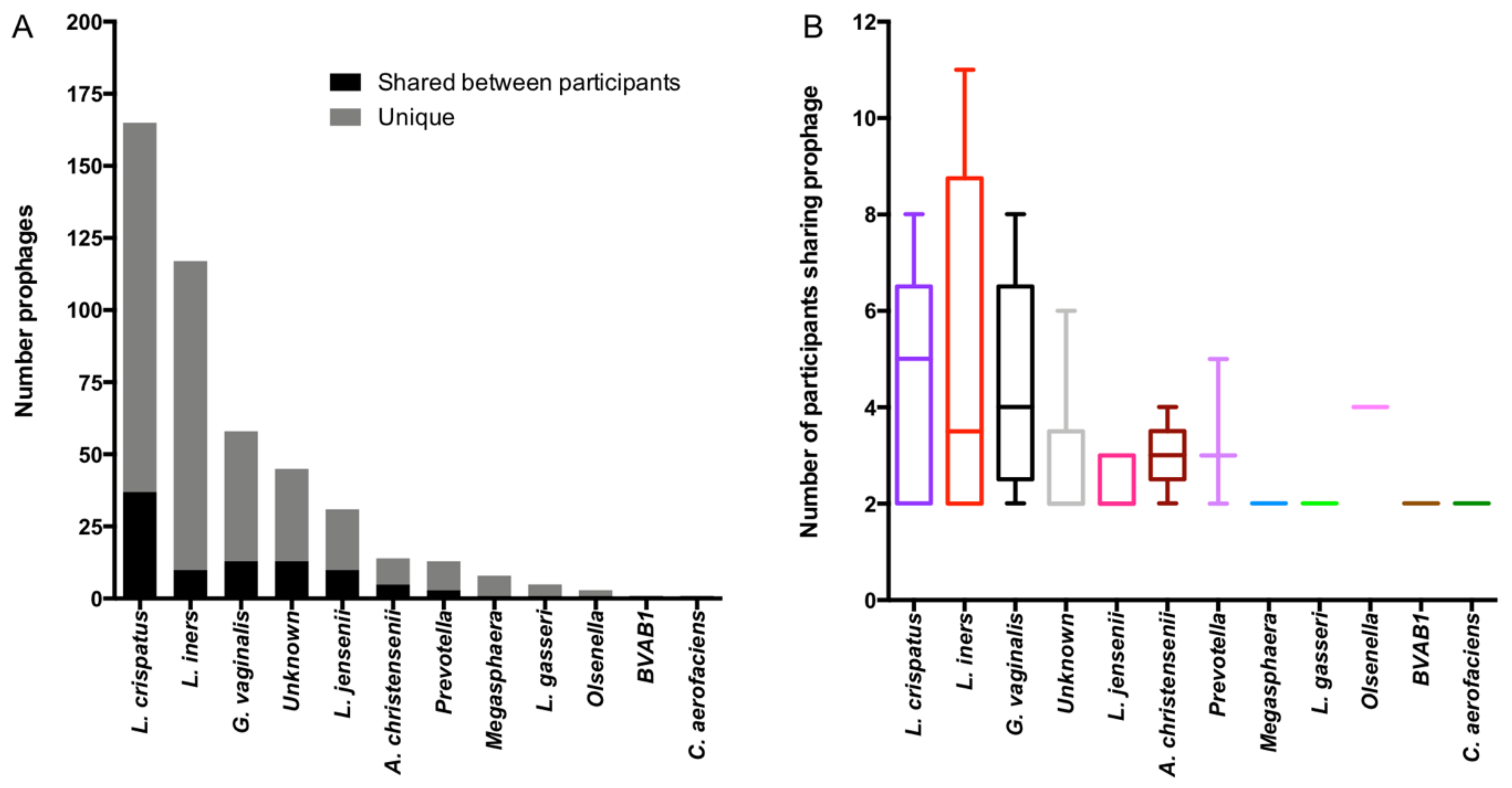
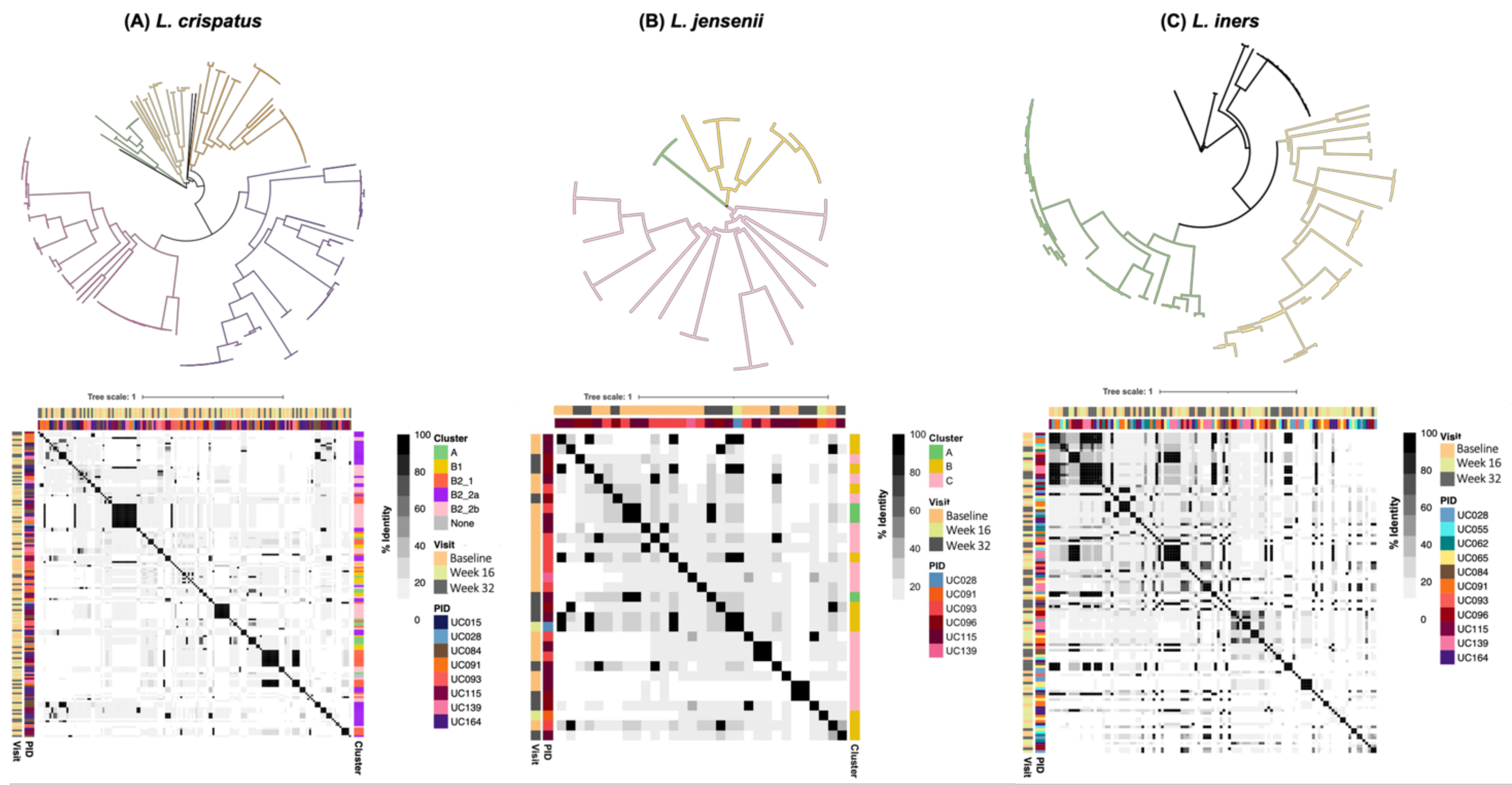
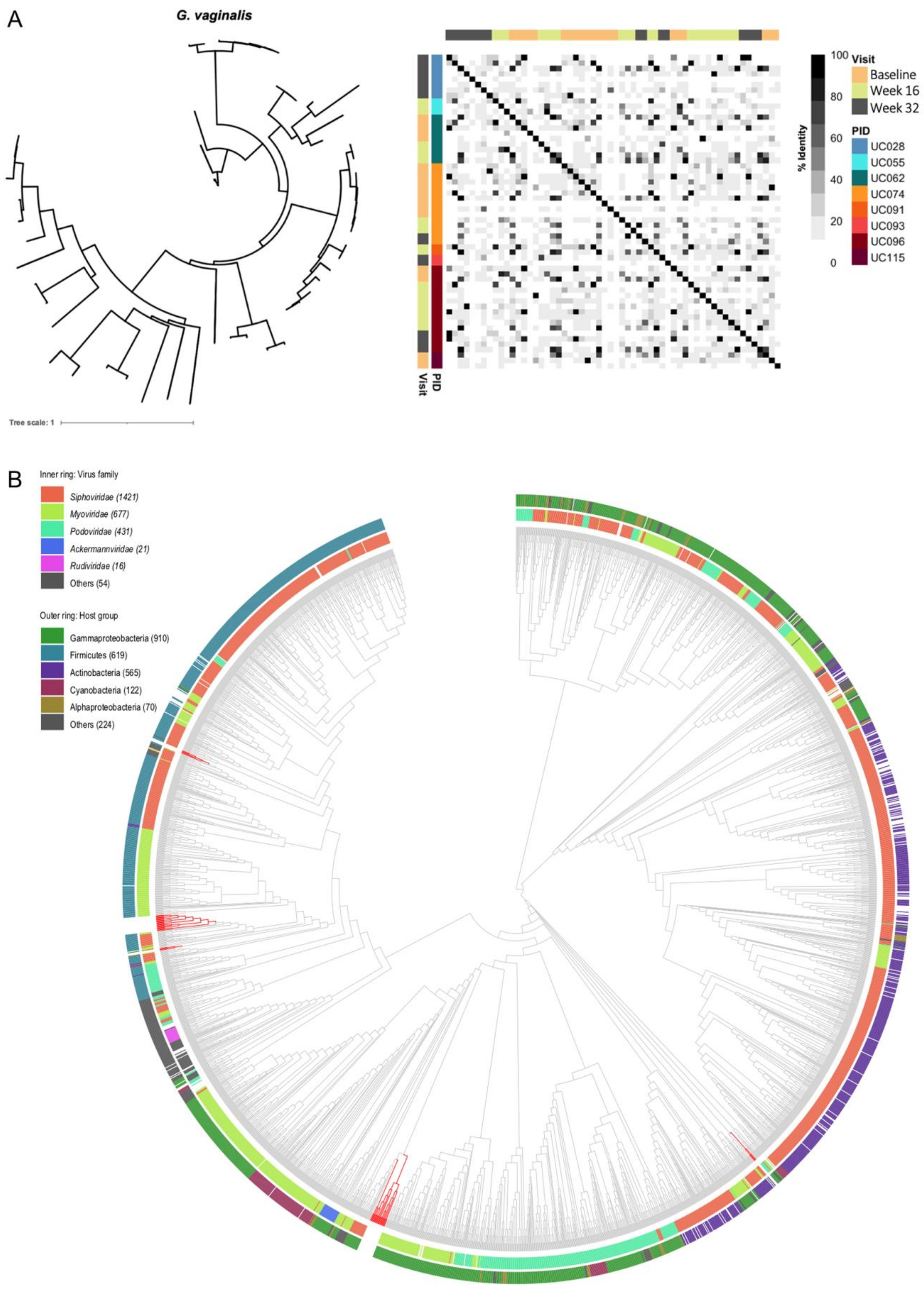
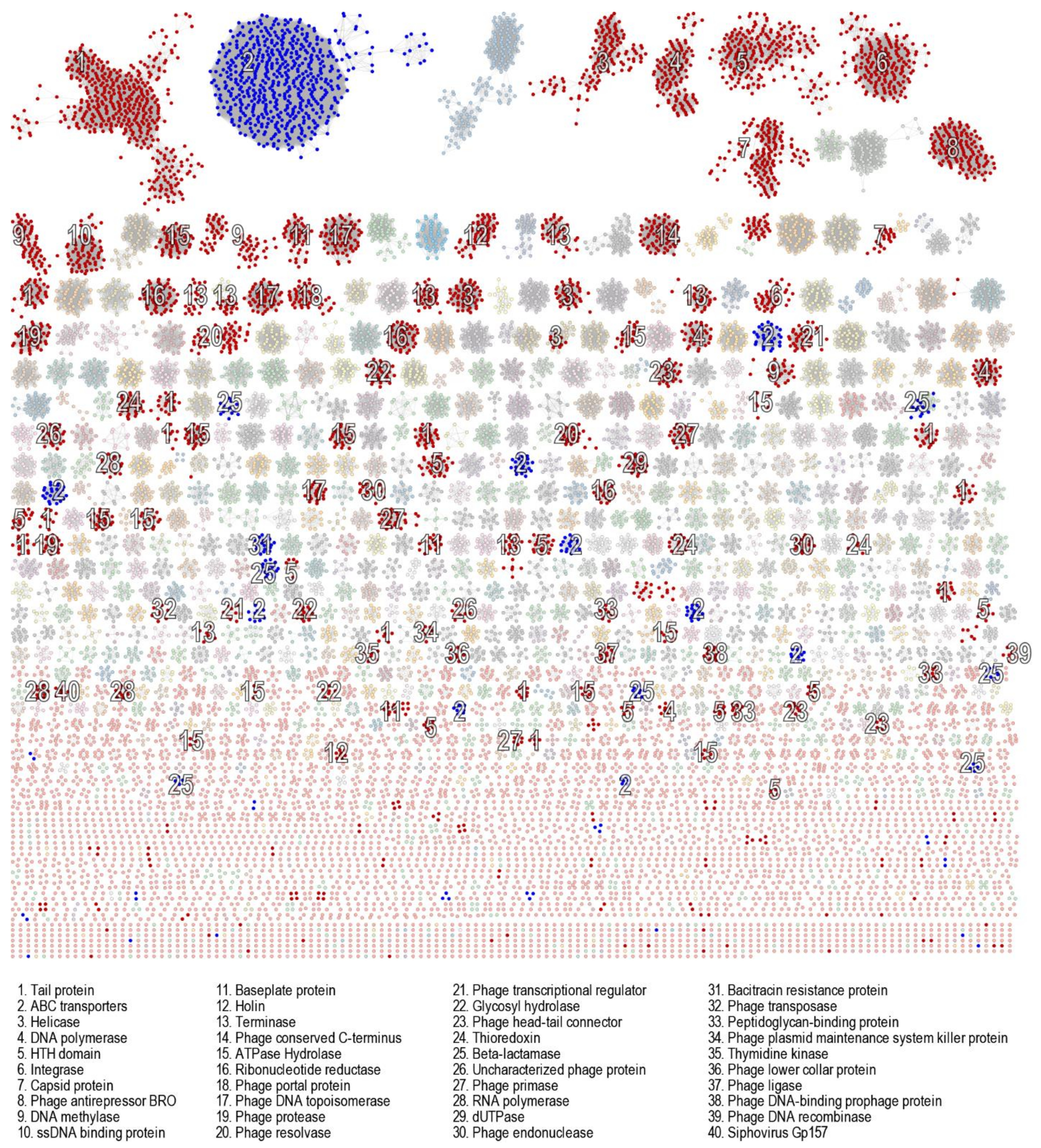
3.6. Genomic Characterisation of Putative Prophages
3.7. Evaluation of Likely Functionality of Putative Prophages
3.8. Evaluation of Antibiotic Resistance Gene Presence in Putative Prophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317. [Google Scholar] [CrossRef]
- Wick, L.M.; Qi, W.; Lacher, D.W.; Whittam, T.S. Evolution of genomic content in the stepwise emergence of Escherichia coli O157:H7. J. Bacteriol. 2005, 187, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abt, M.C.; Buffie, C.G.; Sušac, B.; Becattini, S.; Carter, R.A.; Leiner, I.; Keith, J.W.; Artis, D.; Osborne, L.C.; Pamer, E.G. TLR-7 activation enhances IL-22-mediated colonization resistance against vancomycin-resistant enterococcus. Sci. Transl. Med. 2016, 8, 327ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lysholm, F.; Wetterbom, A.; Lindau, C.; Darban, H.; Bjerkner, A.; Fahlander, K.; Lindberg, A.M.; Persson, B.; Allander, T.; Andersson, B. Characterization of the Viral Microbiome in Patients with Severe Lower Respiratory Tract Infections, Using Metagenomic Sequencing. PLoS ONE 2012, 7, e30875. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.; Abedon, S.T. Classification of Bacteriophages. In The Bacteriophages; Oxford University Press: New York, NY, USA, 2006; pp. 8–11. [Google Scholar]
- Martín, R.; Escobedo, S.; Suárez, J.E. Induction, structural characterization, and genome sequence of Lv1, a prophage from a human vaginal Lactobacillus jensenii strain. Int. Microbiol. 2010, 13, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, S.I.; Tao, L. Induction of vaginal Lactobacillus phages by the cigarette smoke chemical benzo[a]pyrene diol epoxide. Mutat. Res. 2000, 466, 57–62. [Google Scholar] [CrossRef]
- Diard, M.; Bakkeren, E.; Cornuault, J.K.; Moor, K.; Hausmann, A.; Sellin, M.E.; Loverdo, C.; Aertsen, A.; Ackermann, M.; De Paepe, M.; et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science 2017, 355, 1211–1215. [Google Scholar] [CrossRef]
- Lunde, M.; Aastveit, A.H.; Blatny, J.M.; Nes, I.F. Effects of Diverse Environmental Conditions on φLC3 Prophage Stability in Lactococcus lactis. Appl. Environ. Microbiol. 2005, 71, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Shousha, A.; Awaiwanont, N.; Sofka, D.; Smulders, F.J.M.; Paulsen, P.; Szostak, M.P.; Humphrey, T.; Hilbert, F. Bacteriophages Isolated from Chicken Meat and the Horizontal Transfer of Antimicrobial Resistance Genes. Appl. Environ. Microbiol. 2015, 81, 4600–4606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuch, R.; Fischetti, V.A. Detailed genomic analysis of the Wbeta and gamma phages infecting Bacillus anthracis: Implications for evolution of environmental fitness and antibiotic resistance. J. Bacteriol. 2006, 188, 3037–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, P.; Chatterjee, A.; Varsani, A.; Trubl, G. Integrating Viral Metagenomics into an Ecological Framework. Annu. Rev. Virol. 2021, 8, 133–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef] [Green Version]
- Duerkop, B.A.; Clements, C.V.; Rollins, D.; Rodrigues, J.L.M.; Hooper, L.V. A composite bacteriophage alters colonization by an intestinal commensal bacterium. Proc. Natl. Acad. Sci. USA 2012, 109, 17621–17626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucouturier, A.; Chain, F.; Langella, P.; Bidnenko, E. Characterization of a Prophage-Free Derivative Strain of Lactococcus lactis ssp. lactis IL1403 Reveals the Importance of Prophages for Phenotypic Plasticity of the Host. Front. Microbiol. 2018, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ko, K.S. Cryptic prophages in a blaNDM-1-bearing plasmid increase bacterial survival against high NaCl concentration, high and low temperatures, and oxidative and immunological stressors. J. Microbiol. 2020, 58, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Renard, A.; Diene, S.M.; Courtier-Martinez, L.; Gaillard, J.B.; Gbaguidi-Haore, H.; Mereghetti, L.; Quentin, R.; Francois, P.; Van Der Mee-Marquet, N. 12/111phiA Prophage Domestication Is Associated with Autoaggregation and Increased Ability to Produce Biofilm in Streptococcus agalactiae. Microorganisms 2021, 9, 1112. [Google Scholar] [CrossRef] [PubMed]
- Laumay, F.; Corvaglia, A.-R.; Diene, S.M.; Girard, M.; Oechslin, F.; van der Mee-Marquet, N.; Entenza, J.M.; François, P. Temperate Prophages Increase Bacterial Adhesin Expression and Virulence in an Experimental Model of Endocarditis Due to Staphylococcus aureus From the CC398 Lineage. Front. Microbiol. 2019, 10, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandon, S. Why Be Temperate: Lessons from Bacteriophage λ. Trends Microbiol. 2016, 24, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Evolution of Superinfection Immunity in Cluster A Mycobacteriophages. mBio 2019, 10, e00971-19. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Cruz, S.; Parlindungan, E.; Erazo Garzon, A.; Alqarni, M.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Mahony, J. Lysogenization of a Lactococcal Host with Three Distinct Temperate Phages Provides Homologous and Heterologous Phage Resistance. Microorganisms 2020, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [Green Version]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541.e5. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef]
- Benler, S.; Yutin, N.; Antipov, D.; Rayko, M.; Shmakov, S.; Gussow, A.B.; Pevzner, P.; Koonin, E.V. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 2021, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e9. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Hill, C. Shining Light on Human Gut Bacteriophages. Front. Cell. Infect. Microbiol. 2020, 10, 481. [Google Scholar] [CrossRef]
- Moreno-Gallego, J.L.; Chou, S.-P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federici, S.; Nobs, S.P.; Elinav, E. Phages and their potential to modulate the microbiome and immunity. Cell. Mol. Immunol. 2021, 18, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Popescu, M.; Van Belleghem, J.D.; Khosravi, A.; Bollyky, P.L. Bacteriophages and the Immune System. Annu. Rev. Virol. 2021, 8, 415–435. [Google Scholar] [CrossRef]
- Bodner, K.; Melkonian, A.L.; Covert, M.W. The Enemy of My Enemy: New Insights Regarding Bacteriophage-Mammalian Cell Interactions. Trends Microbiol. 2021, 29, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef] [Green Version]
- Calisher, C.H.; Briese, T.; Brister, J.R.; Charrel, R.N.; Dürrwald, R.; Ebihara, H.; Fulhorst, C.F.; Gāo, G.F.; Groschup, M.H.; Haddow, A.D.; et al. Strengthening the Interaction of the Virology Community with the International Committee on Taxonomy of Viruses (ICTV) by Linking Virus Names and Their Abbreviations to Virus Species. Syst. Biol. 2019, 68, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Cortez, M.H.; Dushoff, J.; Weitz, J.S. When to be temperate: On the fitness benefits of lysis vs. lysogeny. Virus Evol. 2020, 6, veaa042. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Sausset, R.; Petit, M.A.; Gaboriau-Routhiau, V.; De Paepe, M. New insights into intestinal phages. Mucosal Immunol. 2020, 13, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Kilic, A.O.; Pavlova, S.I.; Alpay, S.; Kilic, S.S.; Tao, L. Comparative Study of Vaginal Lactobacillus Phages Isolated from Women in the United States and Turkey: Prevalence, Morphology, Host Range, and DNA Homology. Clin. Diagn. Lab. Immunol. 2001, 8, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damelin, L.H.; Paximadis, M.; Mavri-damelin, D.; Birkhead, M.; Lewis, D.A.; Tiemessen, C.T. Identification of predominant culturable vaginal Lactobacillus species and associated bacteriophages from women with and without vaginal discharge syndrome in South Africa. J. Med. Microbiol. 2011, 60, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhangming, P.; Ahmed, S.F.; Xiao, H.; Jianxin, Z.; Hao, Z.; Paul, R.R.; Wenwei, L.; Wei, C.; Ann, M.L.; Dele, O. Comprehensive Scanning of Prophages in Lactobacillus: Distribution, Diversity, Antibiotic Resistance Genes, and Linkages with CRISPR-Cas Systems. mSystems 2021, 6, e01211-20. [Google Scholar] [CrossRef]
- Happel, A.-U.; Kullin, B.; Gamieldien, H.; Wentzel, N.; Zauchenberger, C.Z.; Jaspan, H.B.; Dabee, S.; Barnabas, S.L.; Jaumdally, S.Z.; Dietrich, J.; et al. Exploring potential of vaginal Lactobacillus isolates from South African women for enhancing treatment for bacterial vaginosis. PLoS Pathog. 2020, 16, e1008559. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, R.R.; Haahr, T.; Humaidan, P.; Jensen, J.S.; Kot, W.P.; Castro-Mejia, J.L.; Deng, L.; Leser, T.D.; Nielsen, D.S. Characterization of the Vaginal DNA Virome in Health and Dysbiosis. Viruses 2020, 12, 1143. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.C.; Moron, A.F.; Forney, L.J.; Linhares, I.M.; Sabino, E.; Costa, S.F.; Mendes-Correa, M.C.; Witkin, S.S. Identification of bacteriophages in the vagina of pregnant women: A descriptive study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 976–982. [Google Scholar] [CrossRef]
- Gill, K.; Happel, A.-U.; Pidwell, T.; Mendelsohn, A.; Duyver, M.; Johnson, L.; Meyer, L.; Slack, C.; Strode, A.; Mendel, E.; et al. An open-label, randomized crossover study to evaluate the acceptability and preference for contraceptive options in female adolescents, 15 to 19 years of age in Cape Town, as a proxy for HIV prevention methods (UChoose). J. Int. AIDS Soc. 2020, 23, e25626. [Google Scholar] [CrossRef] [PubMed]
- Balle, C.; Konstantinus, I.N.; Jaumdally, S.Z.; Havyarimana, E.; Lennard, K.; Esra, R.; Barnabas, S.L.; Happel, A.-U.; Moodie, Z.; Gill, K.; et al. Hormonal contraception alters vaginal microbiota and cytokines in South African adolescents in a randomized trial. Nat. Commun. 2020, 11, 5578. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, T.A.K.; Li, P.-E.; Scholz, M.B.; Chain, P.S.G. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Espino, D.; Roux, S.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Huntemann, M.; Reddy, T.B.K.; Pons, J.C.; Llabres, M.; et al. IMG/VR v.2.0: An integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 2019, 47, D678–D686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhou, F.; Gan, R.; Ren, C.; Jia, Y.; Yu, L.; Huang, Z. PHISDetector: A tool to detect diverse in silico phage-host interaction signals for virome studies. bioRxiv 2020, 661074. [Google Scholar] [CrossRef] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 1019–1037. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Iranzo, J.; Krupovic, M.; Koonin, E.V.; Roger, H.; Sergei, M.; Anca, S. The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing. mBio 2021, 7, e00978-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Villion, M.; Moineau, S. Bacteriophages of Lactobacillus. Front. Biosci. 2009, 14, 1661–1683. [Google Scholar] [CrossRef] [Green Version]
- Cantalupo, P.G.; Calgua, B.; Zhao, G.; Hundesa, A.; Wier, A.D.; Katz, J.P.; Grabe, M.; Hendrix, R.W.; Girones, R.; Wang, D.; et al. Raw sewage harbors diverse viral populations. mBio 2011, 2, e00180-11. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.-H.; Lin, X.B.; Zhang, S.; Tollenaar, S.L.; Özçam, M.; Dunphy, C.; Walter, J.; van Pijkeren, J.-P. Prophages in Lactobacillus reuteri Are Associated with Fitness Trade-Offs but Can Increase Competitiveness in the Gut Ecosystem. Appl. Environ. Microbiol. 2019, 86, e01922-19. [Google Scholar] [CrossRef] [Green Version]
- Nanda, A.M.; Thormann, K.; Frunzke, J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 2015, 197, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes-Soares, H.; Suzuki, H.; Hickey, R.J.; Forney, L.J. Comparative Functional Genomics of Lactobacillus spp. Reveals Possible Mechanisms for Specialization of Vaginal Lactobacilli to Their Environment. J. Bacteriol. 2014, 196, 1458–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeoman, C.J.; Yildirim, S.; Thomas, S.M.; Durkin, A.S.; Torralba, M.; Sutton, G.; Buhay, C.J.; Ding, Y.; Dugan-Rocha, S.P.; Muzny, D.M.; et al. Comparative Genomics of Gardnerella vaginalis Strains Reveals Substantial Differences in Metabolic and Virulence Potential. PLoS ONE 2010, 5, e12411. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Ly, M.; Bonilla, N.; Pride, D.T. The human urine virome in association with urinary tract infections. Front. Microbiol. 2015, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garretto, A.; Miller-Ensminger, T.; Wolfe, A.J.; Putonti, C. Bacteriophages of the lower urinary tract. Nat. Rev. Urol. 2019, 16, 422–432. [Google Scholar] [CrossRef]
- Barylski, J.; Kropinski, A.M.; Alikhan, N.-F.; Adriaenssens, E.M.; Consortium, I.R. ICTV Virus Taxonomy Profile: Herelleviridae. J. Gen. Virol. 2020, 101, 362–363. [Google Scholar] [CrossRef]
- Knezevic, P.; Adriaenssens, E.M.; Consortium, I.R. ICTV Virus Taxonomy Profile: Inoviridae. J. Gen. Virol. 2021, 102, 001614. [Google Scholar] [CrossRef]
- Malki, K.; Shapiro, J.W.; Price, T.K.; Hilt, E.E.; Thomas-White, K.; Sircar, T.; Rosenfeld, A.B.; Kuffel, G.; Zilliox, M.J.; Wolfe, A.J.; et al. Genomes of Gardnerella Strains Reveal an Abundance of Prophages within the Bladder Microbiome. PLoS ONE 2016, 11, e0166757. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Janulaitiene, M.; Paliulyte, V.; Grinceviciene, S.; Zakareviciene, J.; Vladisauskiene, A.; Marcinkute, A.; Pleckaityte, M. Prevalence and distribution of Gardnerella vaginalis subgroups in women with and without bacterial vaginosis. BMC Infect. Dis. 2017, 17, 394. [Google Scholar] [CrossRef]
- Cornejo, O.E.; Hickey, R.J.; Suzuki, H.; Forney, L.J. Focusing the diversity of Gardnerella vaginalis through the lens of ecotypes. Evol. Appl. 2017, 11, 312–324. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Hakim, J.A.; Crossman, D.K.; Lefkowitz, E.J.; Morrow, C.D. Sharing of gut microbial strains between selected individual sets of twins cohabitating for decades. PLoS ONE 2019, 14, e0226111. [Google Scholar] [CrossRef] [Green Version]
- Marrazzo, J.M.; Antonio, M.; Agnew, K.; Hillier, S.L. Distribution of Genital Lactobacillus Strains Shared by Female Sex Partners. J. Infect. Dis. 2009, 199, 680–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, A.L. Vaginal bacterial phaginosis? Sex. Trans. Infect. 1999, 75, 352–353. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline n = 13 | Week 16 n = 13 | Week 32 n = 12 | |
|---|---|---|---|
| Age, median years (IQR) | 16 (16, 17) | - | - |
| BMI, median (IQR) | 23.6 (22.0, 25.1) | 23.9 (22.5, 24.9) | 24.3 (23.5, 26.3) |
| BV prevalence, n (%) | |||
| BV+ (Nugent 7–10) | 8 (61.5) | 8 (61.5) | 8 (66.7) |
| BV intermediate (Nugent 4–6) | 3 (23.1) | 1 (7.7) | 0 (0.0) |
| BV− (Nugent 0–3) | 2 (15.4) | 4 (30.8) | 4 (33.3) |
| Yeast prevalence (Gram stain), n (%) | 2 (15.4) | 0 (0.0) | 2 (16.7) |
| HSV-2 serology prevalence, n (%) | 2 (15.4) | 2 (15.4) | 2 (16.7) |
| CST distribution, n (%) a | |||
| CST-I | 4 (30.8) | 5 (38.5) | 4 (33.3) |
| CST-III | 5 (38.5) | 5 (38.5) | 5 (41.7) |
| CST-IV | 3 (23.1) | 3 (23.1) | 3 (25.0) |
| Shannon Index, median (IQR) a | 0.71 (0.38, 1.34) | 1.49 (0.39, 1.75) | 1.12 (0.89, 1.50) |
| Days since the last menstrual period, median (IQR) | 66 (23, 93) | 19 (12, 54) | 19 (10, 21) |
| Age menarche, median (IQR) | 13 (13, 14) | - | - |
| Previously pregnant, n (%) | 1 (7.7) | - | - |
| Hormonal contraception at time of sampling, n (%) | |||
| None | 1 (7.7) | 0 (0.0) | 0 (0.0) |
| Injectables (NET-EN/DMPA) | 12 (92.3) | 3 (23.1) | 5 (41.7) |
| COCP | 0 (0.0) | 5 (38.5) | 2 (16.7) |
| CCVR | 0 (0.0) | 5 (38.5) | 5 (41.7) |
| Sexual risk behaviour | |||
| Age of sexual debut, median [IQR] | 15 [14, 16] | - | - |
| Any sexual partner(s) past year, n (%) | 11 (91.7) | - | - |
| Multiple sexual partners past year, n (%) | 1 (7.7) | - | - |
| New partner past year, n (%) | 4 (33.3) | - | - |
| Condom use during last penile-vaginal intercourse, n (%) | 9 (75.0) | 7 (70.0) | 9 (75.0) |
| Reported alcohol use, n (%) | 2 (15.4) | 2 (15.4) | 4 (33.3) |
| Reported cannabis use, n (%) | 1 (7.7) | 0 (0) | 0 (0) |
| Family | Genus | Species (Number of Adolescents in Which Identified) |
|---|---|---|
| Siphoviridae | Unclassified | Stx2-converting phage 1717 (1) |
| Streptococcus phage phiARI0468-2 (2) | ||
| Streptococcus phage phiARI0462 (1) | ||
| Streptococcus phage phiARI0031 (1) | ||
| Streptococcus phage PH10 (3) | ||
| Streptococcus phage MM1 (1) | ||
| Streptococcus phage Dp-1 (2) | ||
| Streptococcus phage 5093 (1) | ||
| Enterococcus phage vB_EfaS_IME197 (1) | ||
| Lactococcus phage WRP3 (1) | ||
| Lactococcus phage Q54 (1) | ||
| Lactococcus phage bIL311 (2) | ||
| Lactobacillus phage PLE2 (1) | ||
| Lactobacillus phage phiJB (1) | ||
| Cronobacter phage ENT39118 (1) | ||
| Clostridium phage phiCP39-O (2) | ||
| Clostridium phage phiCD211 (2) | ||
| Brevibacillus phage Sundance (1) | ||
| Bacillus phage vB_BtS_BMBtp3 (1) | ||
| Bacillus phage vB_BanS-Tsamsa (1) | ||
| Andromedavirus | Bacillus virus Blastoid (1) | |
| Ceetrepovirus | Corynebacterium virus Zion (7) | |
| Doucettevirus | Propionibacterium phage E6 (2) | |
| Magadivirus | Bacillus phage Mgbh1 (1) | |
| Moineauvirus | Streptococcus virus Sfi19 (2) | |
| Streptococcus virus phiAbc2 (1) | ||
| Poushouvirus | Corynebacterium phage Poushou (1) | |
| Sextaecvirus | Staphylococcus phage 6ec (1) | |
| Spbetavirus | Bacillus virus SPbeta (1) | |
| Myoviridae | Unclassified | Bacillus virus G (4) |
| Enterobacteria phage phi92 (1) | ||
| Shigella phage SfIV (2) | ||
| Sphingomonas phage PAU (3) | ||
| Streptococcus phage EJ-1 (2) | ||
| Abouovirus | Brevibacillus phage Abouo (1) | |
| Firehammervirus | Campylobacter virus CP21 (1) | |
| Campylobacter virus CPt10 (1) | ||
| Peduovirinae | Pseudomonas phage phi3 (1) | |
| Escherichia phage pro147 (1) | ||
| Punavirus | Escherichia virus P1 (1) | |
| Salmonella phage SJ46 (1) | ||
| Vequintavirinae | Klebsiella phage vB_KpnM_KB57 (1) | |
| Podoviridae | Lederbergvirus | Salmonella phage vB_SemP_Emek (1) |
| Picovirinae | Streptococcus phage Cp1 (2) | |
| Enterococcus phage EF62phi (1) | ||
| Uetakevirus | Escherichia phage TL-2011b | |
| Inoviridae | Unclassified | Propionibacterium phage B5 (1) |
| Herelleviridae | Unclassified | Lactobacillus virus Lb338-1 (1) |
| Brochothrix phage A9 (1) | ||
| Bastillevirinae | Bacillus phage Deep Blue (3) | |
| Brockvirinae | Enterococcus phage EFDG1 (3) | |
| Spounavirinae | Bacillus virus SPO1 (1) | |
| Bacillus phage Shanette (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Happel, A.-U.; Balle, C.; Maust, B.S.; Konstantinus, I.N.; Gill, K.; Bekker, L.-G.; Froissart, R.; Passmore, J.-A.; Karaoz, U.; Varsani, A.; et al. Presence and Persistence of Putative Lytic and Temperate Bacteriophages in Vaginal Metagenomes from South African Adolescents. Viruses 2021, 13, 2341. https://doi.org/10.3390/v13122341
Happel A-U, Balle C, Maust BS, Konstantinus IN, Gill K, Bekker L-G, Froissart R, Passmore J-A, Karaoz U, Varsani A, et al. Presence and Persistence of Putative Lytic and Temperate Bacteriophages in Vaginal Metagenomes from South African Adolescents. Viruses. 2021; 13(12):2341. https://doi.org/10.3390/v13122341
Chicago/Turabian StyleHappel, Anna-Ursula, Christina Balle, Brandon S. Maust, Iyaloo N. Konstantinus, Katherine Gill, Linda-Gail Bekker, Rémy Froissart, Jo-Ann Passmore, Ulas Karaoz, Arvind Varsani, and et al. 2021. "Presence and Persistence of Putative Lytic and Temperate Bacteriophages in Vaginal Metagenomes from South African Adolescents" Viruses 13, no. 12: 2341. https://doi.org/10.3390/v13122341
APA StyleHappel, A.-U., Balle, C., Maust, B. S., Konstantinus, I. N., Gill, K., Bekker, L.-G., Froissart, R., Passmore, J.-A., Karaoz, U., Varsani, A., & Jaspan, H. (2021). Presence and Persistence of Putative Lytic and Temperate Bacteriophages in Vaginal Metagenomes from South African Adolescents. Viruses, 13(12), 2341. https://doi.org/10.3390/v13122341