
Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage


Abstract
:1. Introduction
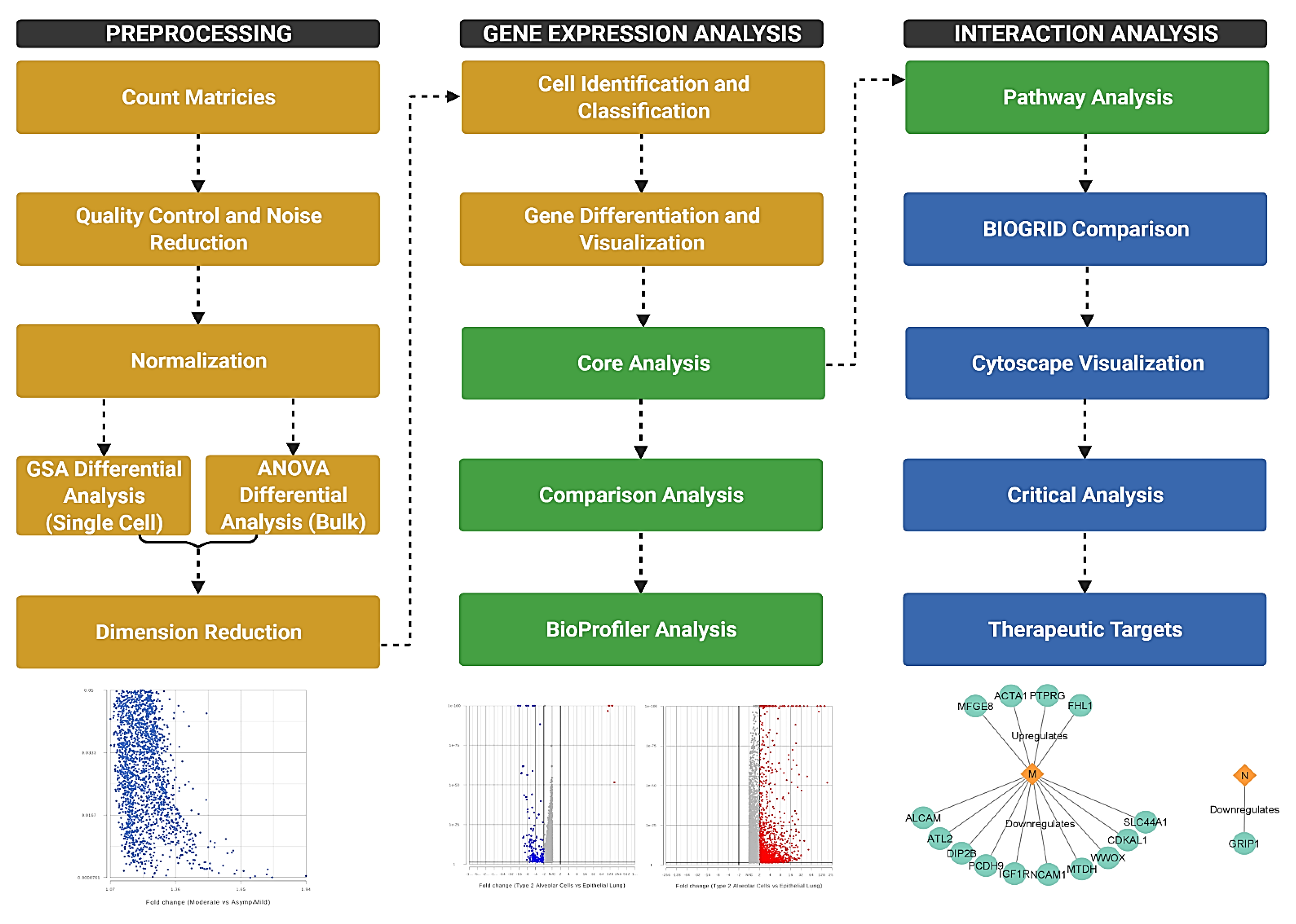
2. Materials and Methods
3. Results and Discussion
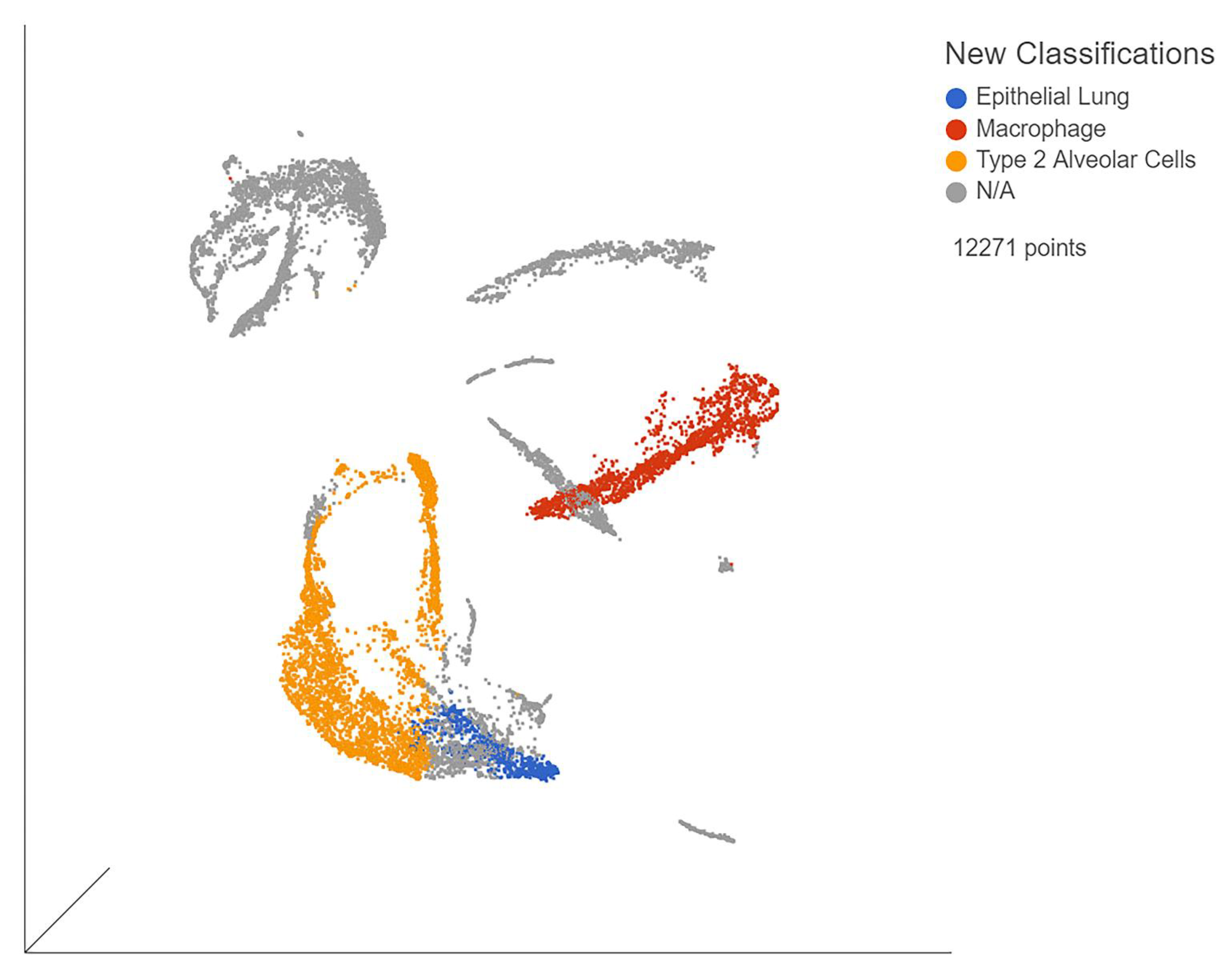
3.1. Single-Cell Immune Profiling Showed Viral Effects per Organ
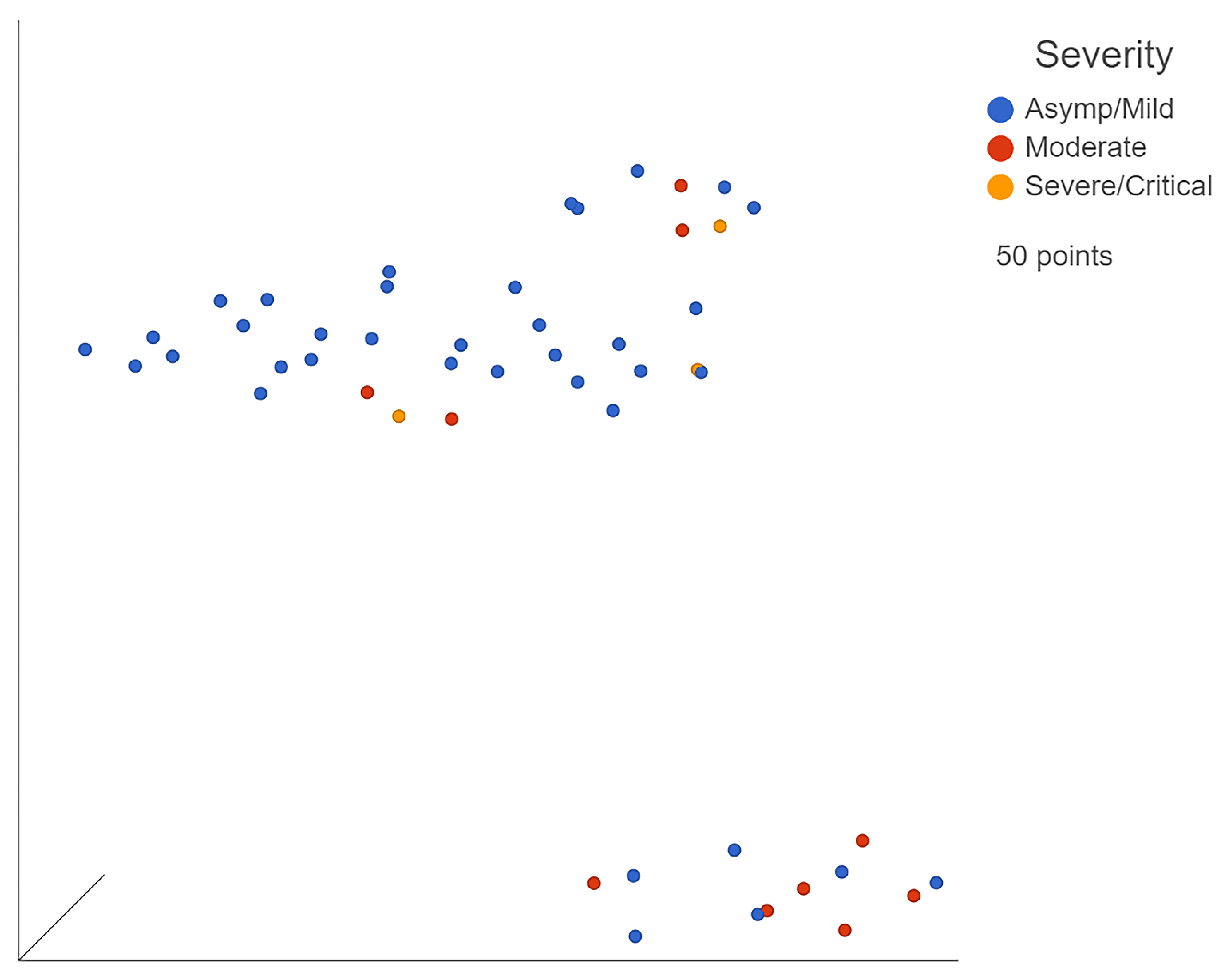
3.2. Bulk RNA Sequencing Identified Differences between Severity Levels
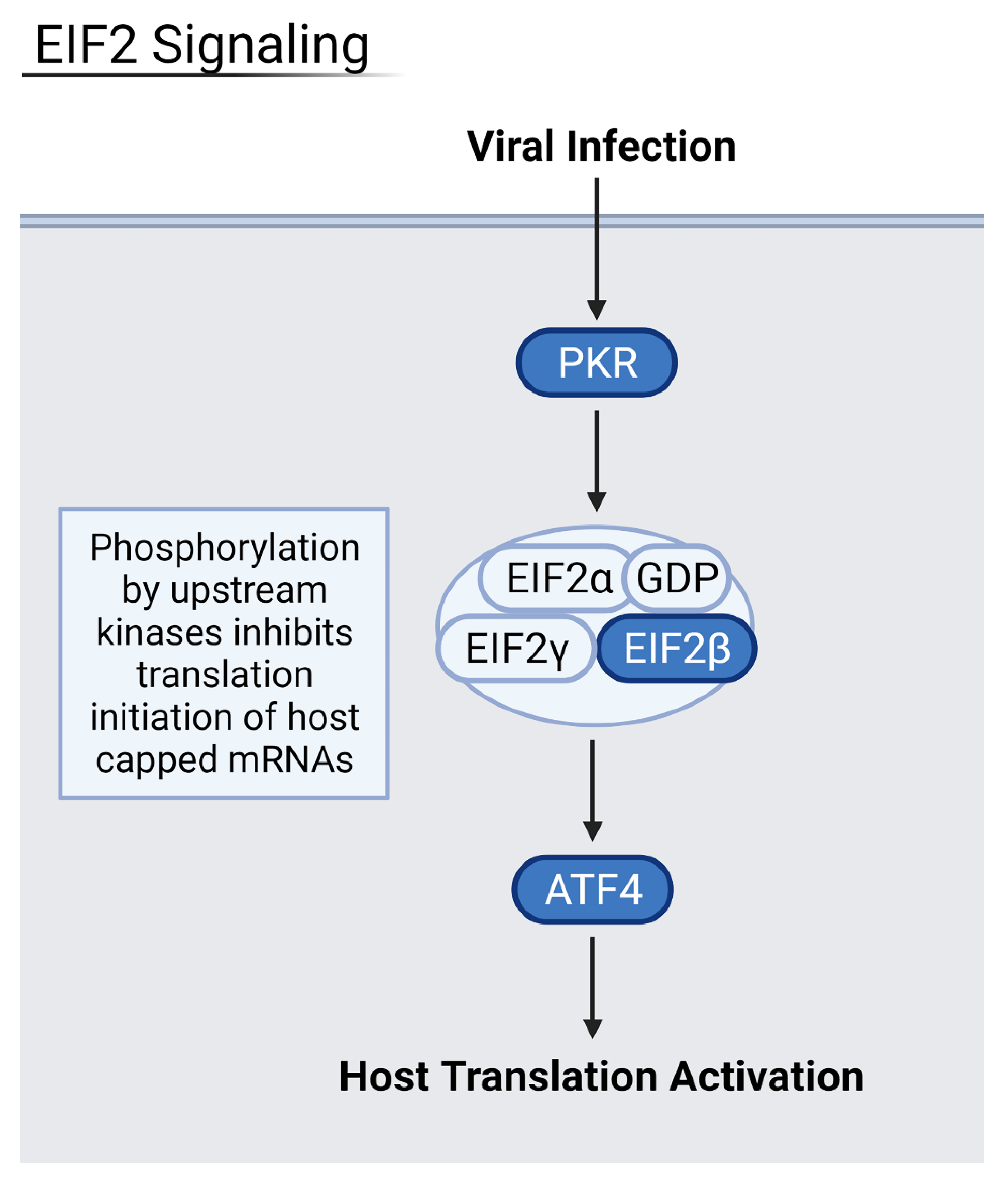
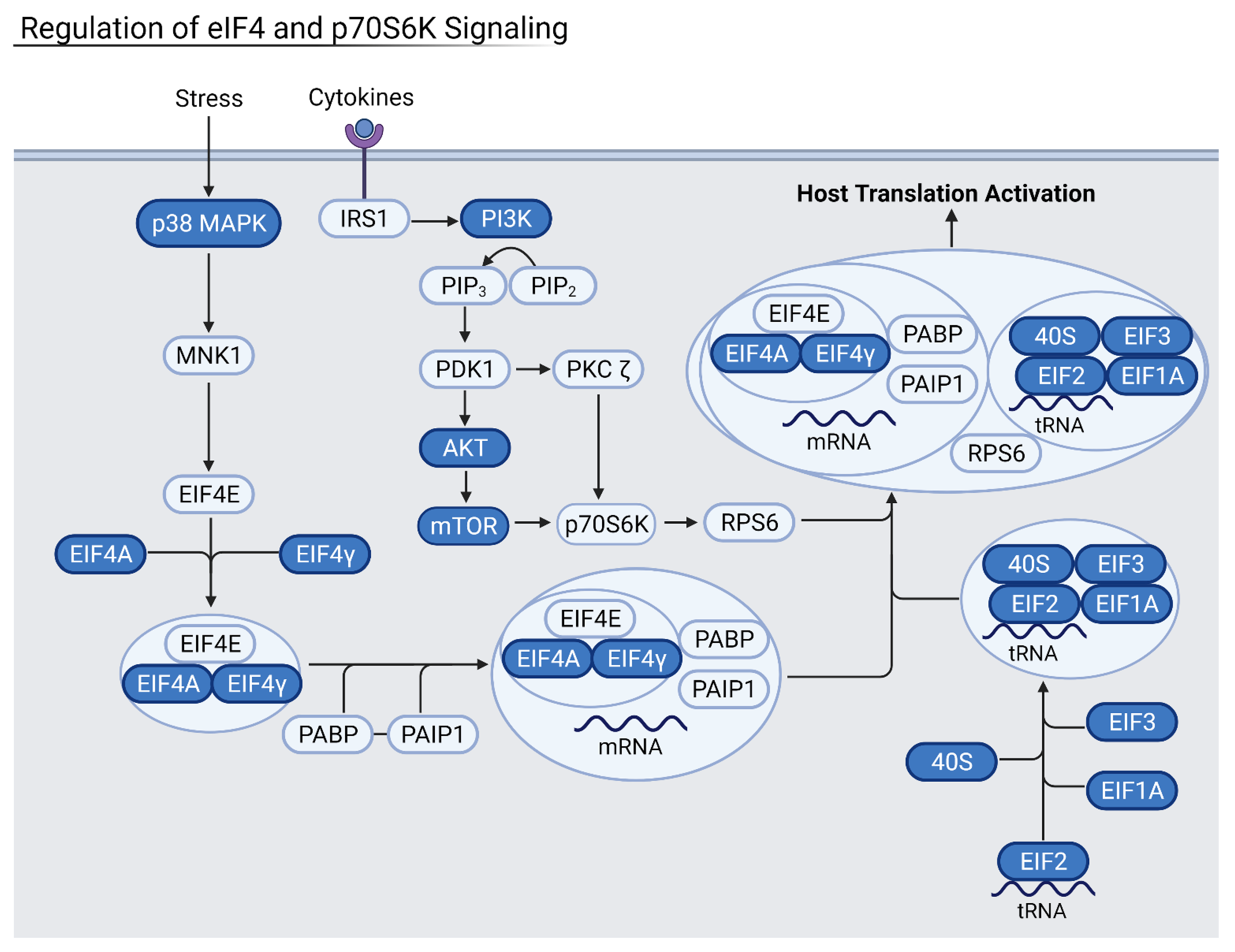
3.3. Viral Disruption of Host Translation and Related Pathways Contributes to Increased Severity
3.4. Reduction of Eukaryotic Initiation Factors Leads to Increased Severity
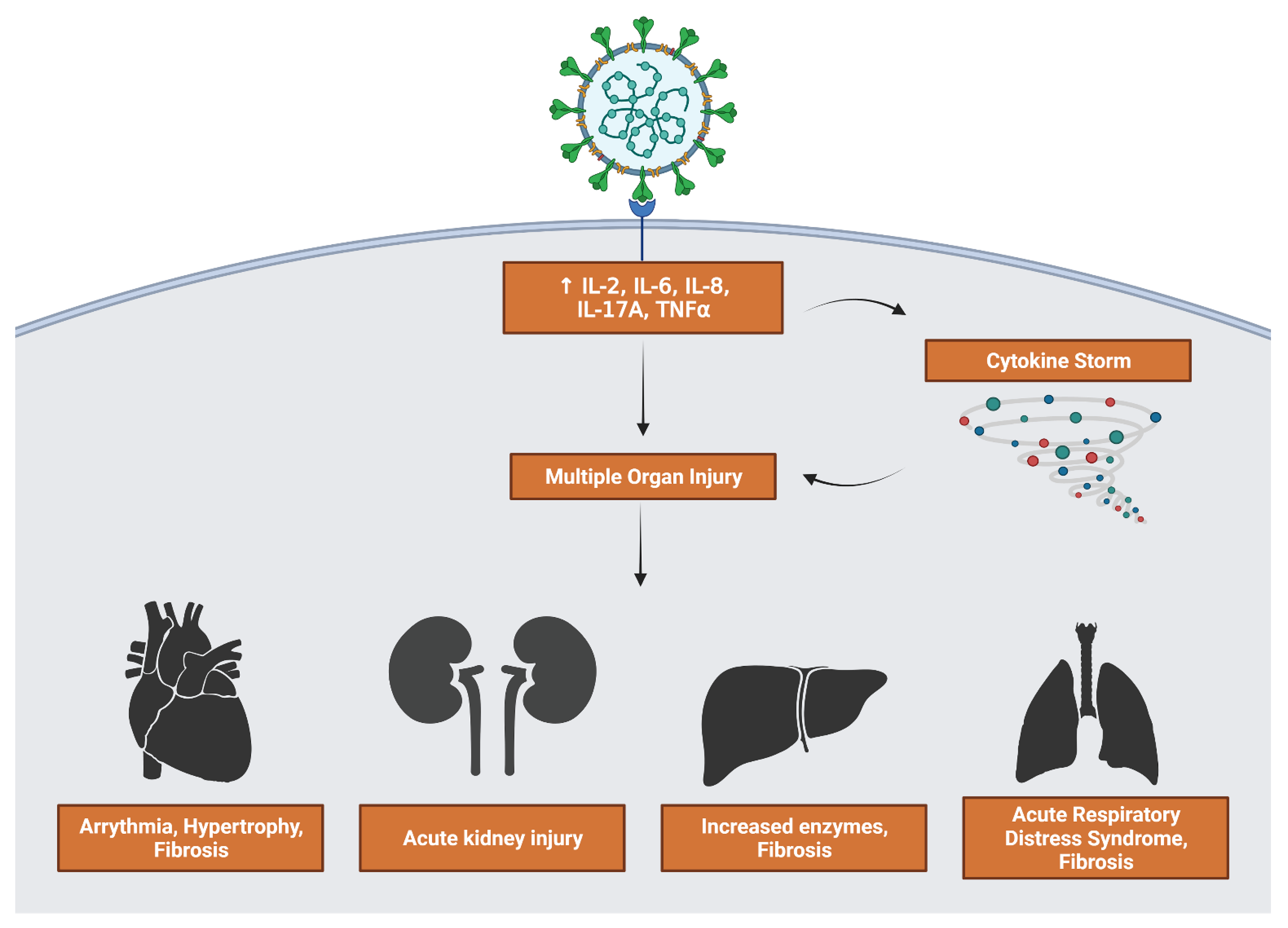
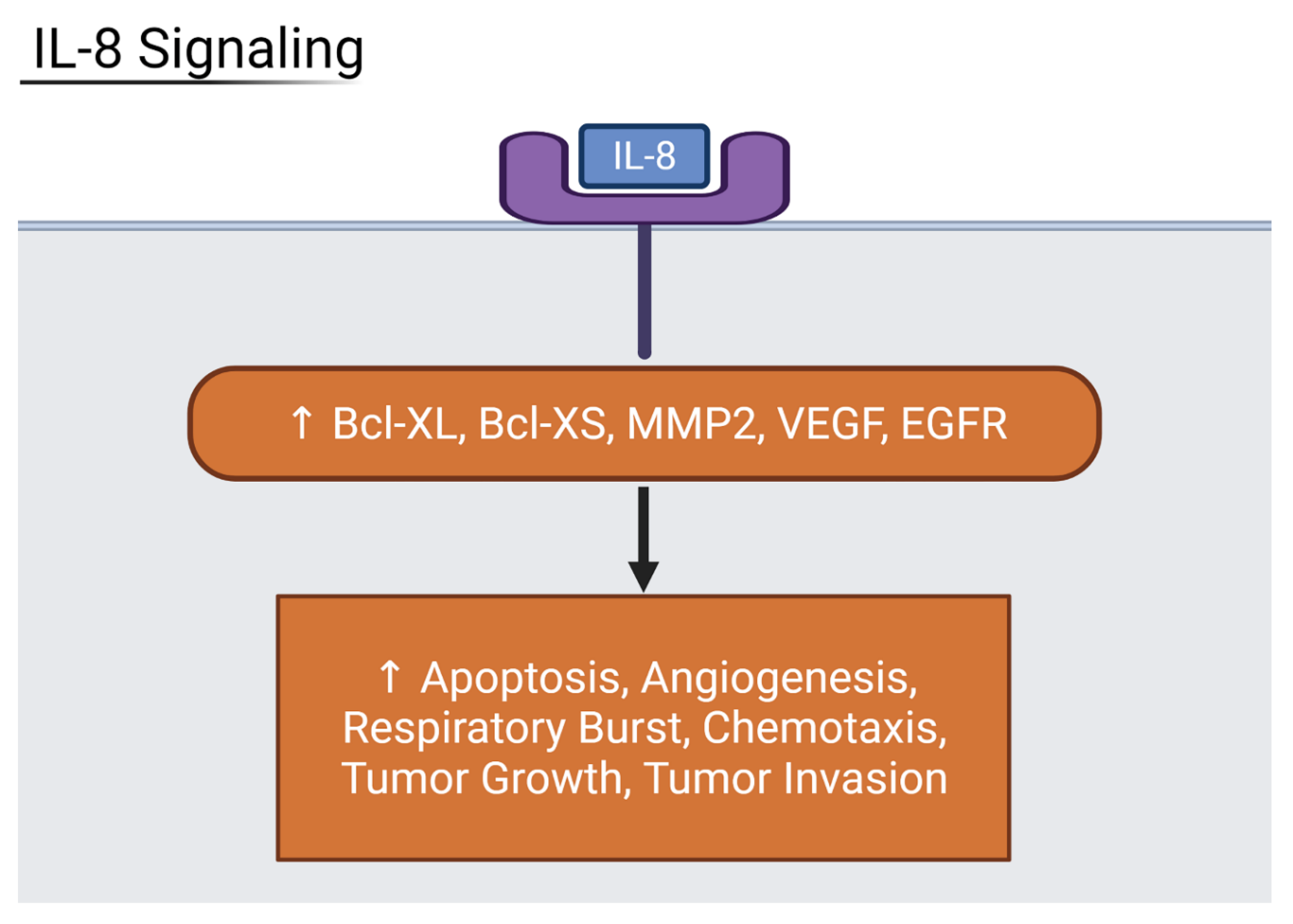
3.5. Inflammatory Interleukin Levels Increase with Severity
3.6. Suppression of Host Translation Leads to Increased Inflammation
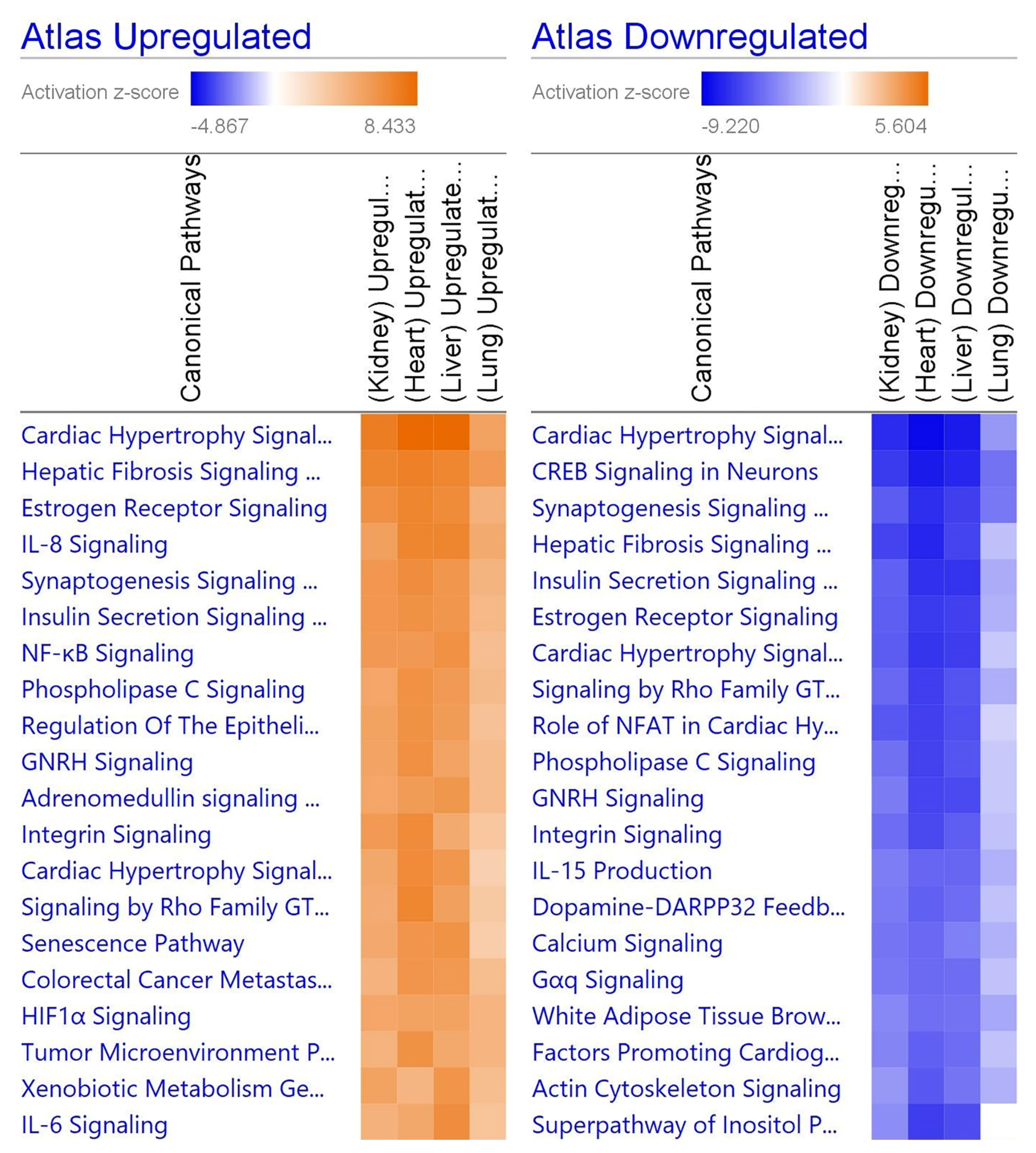
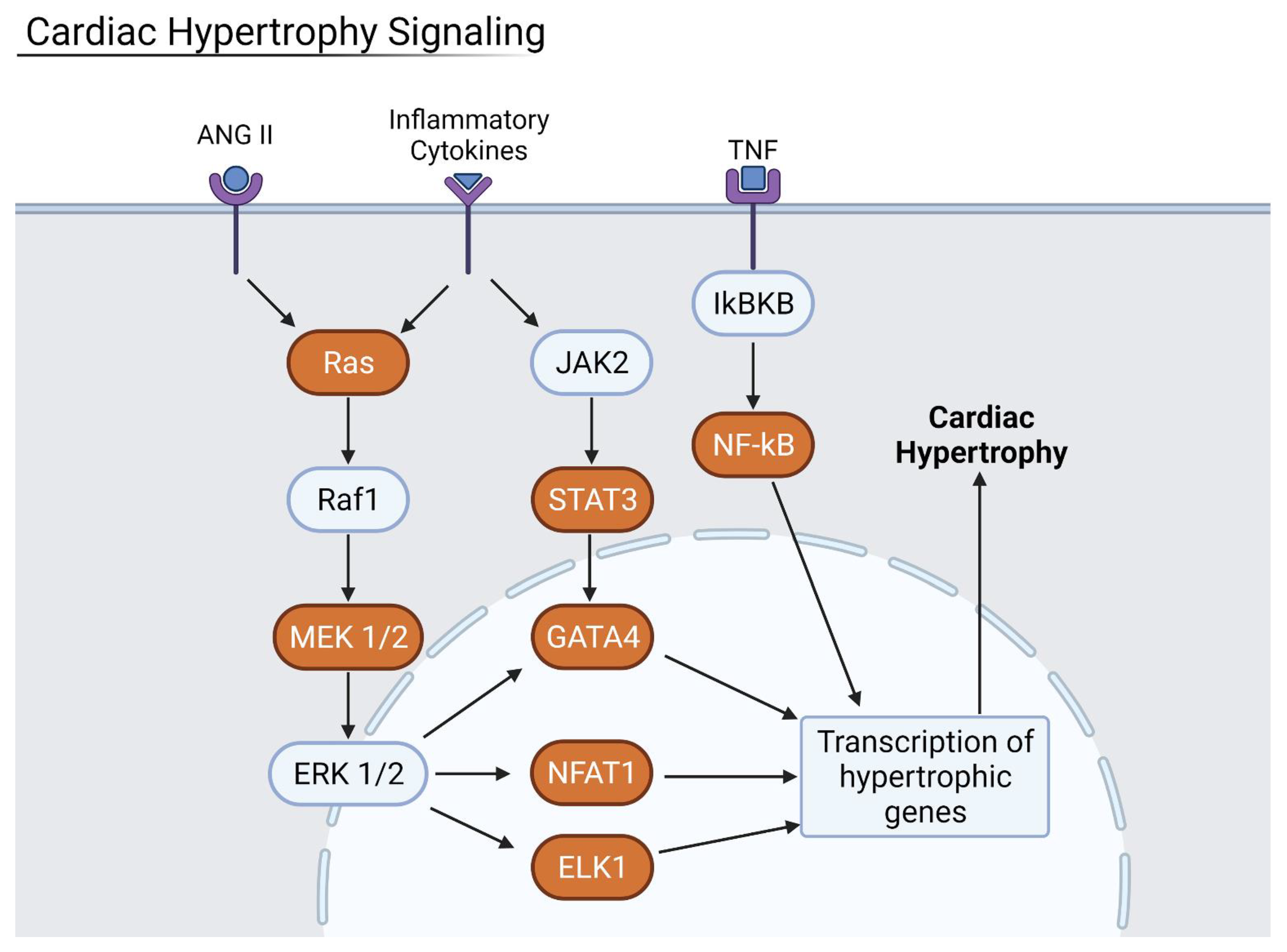
3.7. Inflammation Results in Hypertrophy and Fibrosis
3.8. Interaction Mapping Shows Viral Proteins Disrupt Host Translation
3.9. Commonly Altered Host Genes Correlate with Disease Symptoms
3.10. Individual Organs Have Unique Viral Interactions
3.11. Potential Therapeutic Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.; Pranata, R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): Systematic review and meta-analysis. J. Intensive Care 2020, 8, 36. [Google Scholar] [CrossRef]
- Burgos-Blasco, B.; Güemes-Villahoz, N.; Santiago, J.L.; Fernandez-Vigo, J.I.; Espino-Paisán, L.; Sarriá, B.; García-Feijoo, J.; Martinez-De-La-Casa, J.M. Hypercytokinemia in COVID-19: Tear cytokine profile in hospitalized COVID-19 patients. Exp. Eye Res. 2020, 200, 108253. [Google Scholar] [CrossRef] [PubMed]
- Matsuishi, Y.; Mathis, B.; Shimojo, N.; Subrina, J.; Okubo, N.; Inoue, Y. Severe COVID-19 Infection Associated with Endothelial Dysfunction Induces Multiple Organ Dysfunction: A Review of Therapeutic Interventions. Biomedicines 2021, 9, 279. [Google Scholar] [CrossRef]
- Bian, X.-W.; Yao, X.-H.; Ping, Y.-F.; Yu, S.; Shi, Y.; Luo, T.; He, Z.-C.; Tang, R.; Chen, C.; Fu, W.-J.; et al. Autopsy of COVID-19 patients in China. Natl. Sci. Rev. 2020, 7, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Ratho, R.; Kumar, P.; Bhatia, S.; Bora, I.; Mohi, G.; Saxena, S.; Devi, M.; Yadav, D.; Mehariya, S. Multi-Organ Involvement in COVID-19: Beyond Pulmonary Manifestations. J. Clin. Med. 2021, 10, 446. [Google Scholar] [CrossRef]
- Sun, J.; Aghemo, A.; Forner, A.; Valenti, L. COVID-19 and liver disease. Liver Int. 2020, 40, 1278–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayoun, A.A.; Loney, T.; Khansaheb, H.; Ramaswamy, S.; Harilal, D.; Deesi, Z.O.; Varghese, R.M.; Al Suwaidi, H.; Alkhajeh, A.; AlDabal, L.M.; et al. Multiple early introductions of SARS-CoV-2 into a global travel hub in the Middle East. Sci. Rep. 2020, 10, 17720. [Google Scholar] [CrossRef]
- He, X.; Cheng, X.; Feng, X.; Wan, H.; Chen, S.; Xiong, M. Clinical Symptom Differences Between Mild and Severe COVID-19 Patients in China: A Meta-Analysis. Front. Public Health 2021, 8, 561264. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Perrotta, F.; Polito, R.; D’Agnano, V.; Scialò, F.; Bianco, A.; Daniele, A. Metabolic Perturbations and Severe COVID-19 Disease: Implication of Molecular Pathways. Int. J. Endocrinol. 2020, 2020, 8896536. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nat. Cell Biol. 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Misharin, A.; Lu, Z.; Markov, N.; Wunderink, R. Single Cell RNA-Seq Analysis of Bronchial Epithelium in Patients with Severe SARS-CoV-2 Pneumonia. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168215 (accessed on 4 November 2021).
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef]
- Jain, R.; Ramaswamy, S.; Harilal, D.; Uddin, M.; Loney, T.; Nowotny, N.; Alsuwaidi, H.; Varghese, R.; Deesi, Z.; Alkhajeh, A.; et al. Host transcriptomic profiling of COVID-19 patients with mild, moderate, and severe clinical outcomes. Comput. Struct. Biotechnol. J. 2021, 19, 153–160. [Google Scholar] [CrossRef]
- Mishima, Y.; Sonoyama, H.; Ishihara, S.; Oshima, N.; Moriyama, I.; Kawashima, K.; Kinoshita, Y. Interleukin-33 delays recovery of mucosal inflammation via downregulation of homeostatic ABCG5/8 in the colon. Lab. Investig. 2020, 100, 491–502. [Google Scholar] [CrossRef]
- Khezri, M.R. PI3K/AKT signaling pathway: A possible target for adjuvant therapy in COVID-19. Hum. Cell 2021, 34, 700–701. [Google Scholar] [CrossRef]
- Shrestha, N.; Bahnan, W.; Wiley, D.J.; Barber, G.; Fields, K.A.; Schesser, K. Eukaryotic Initiation Factor 2 (eIF2) Signaling Regulates Proinflammatory Cytokine Expression and Bacterial Invasion. J. Biol. Chem. 2012, 287, 28738–28744. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. The role of host eIF2α in viral infection. Virol. J. 2020, 17, 112. [Google Scholar] [CrossRef]
- Zhu, M.-E.; Wang, Q.; Zhou, S.; Wang, B.; Ke, L.; He, P. Recombinant interleukin-2 stimulates lymphocyte recovery in patients with severe COVID-19. Exp. Ther. Med. 2021, 21, 1. [Google Scholar] [CrossRef]
- Shibabaw, T. Inflammatory Cytokine: IL-17A Signaling Pathway in Patients Present with COVID-19 and Current Treatment Strategy. J. Inflamm. Res. 2020, 13, 673–680. [Google Scholar] [CrossRef]
- Wang, G.; Wu, C.; Zhang, Q.; Wu, F.; Yu, B.; Lv, J.; Li, Y.; Li, T.; Zhang, S.; Wu, C.; et al. C-Reactive Protein Level May Predict the Risk of COVID-19 Aggravation. Open Forum Infect. Dis. 2020, 7, ofaa153. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Gao, M.; Fan, H.; Wang, Y.; Xu, X.; Chen, C.; Liu, J.; Kim, J.; Aliyari, R.; et al. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front. Immunol. 2021, 11, 602395. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Vélia, E.; Mavingui, P.; et al. Altered high-density lipoprotein composition and functions during severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef]
- Marie-Laure, F.; Roland, M.; Johannes, Z.; Minh-Ha, N.; Marie, D.; Louis, B.; Laura, C.; Marti, N.-P.; Lauriane, L.; Beat, H.M.; et al. SARS-CoV-2 ORF7b: Is a bat virus protein homologue a major cause of COVID-19 symptoms? bioRxiv 2021. [Google Scholar] [CrossRef]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Filgueira, L.M.; Cervantes, J.B.; Lovelle, O.A.; Herrera, C.; Figueredo, C.; Caballero, J.A.; Sánchez, N.; Berrio, J.; Lorenzo, G.; Cepeda, M.; et al. An anti-CD6 antibody for the treatment of COVID-19 patients with cytokine-release syndrome: Report of three cases. Immunotherapy 2021, 13, 289–295. [Google Scholar] [CrossRef]
- Conca, W.; Alabdely, M.; Albaiz, F.; Foster, M.W.; Alamri, M.; Alkaff, M.; Al-Mohanna, F.; Nagelkerke, N.; Almaghrabi, R.S. Serum β2-microglobulin levels in Coronavirus disease 2019 (COVID-19): Another prognosticator of disease severity? PLoS ONE 2021, 16, e0247758. [Google Scholar] [CrossRef]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507. [Google Scholar] [CrossRef]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform. Biol. Insights 2015, 9s1. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Study Design | Sample Source | Sample Size | Number of COVID Patients | Number of Healthy Control | Type |
|---|---|---|---|---|---|---|
| GSE171668 [12] | COVID-19 autopsy biobank of 420 autopsy specimens, spanning 11 organs and 17 donors. | Various organs (lung, kidney, liver, heart) | 118 | 17 | 0 | Single-cell RNA-seq |
| GSE168215 [13] | Bronchial brushings from 4 patients with COVID-19 were obtained at Northwestern Memorial ICU. | Lung | 9 | 4 | 0 | Single-cell RNA-seq |
| GSE150316 [14] | Autopsy samples from patients deceased due to SARS-CoV-2 infection were collected for total RNA-seq analysis to assess viral load and immune response. | Lung, liver, heart, jejunum, bowel, kidney, placenta | 88 | 5 | 5 samples (patient # not indicated) | Bulk RNA-seq |
| GSE162835 [15] | Shotgun transcriptome sequencing of human RNA obtained from nasopharyngeal swabs of patients with COVID-19. | Nasopharyngeal swabs | 50 | 50 | 0 | Bulk RNA-seq |
| Factors of Severity | Genes and Function |
|---|---|
| Decreased Immune Response | HLA-A, -B, -C—immune regulatory genes NCAM1—decreased immune cell expansion and interferon signaling |
| Factors that Correlate with Severity | APOB—decreased apolipoproteins COVID-19 patient plasma B2M—decreased organ expression contributes to renal and cardiovascular damage |
| Fibrosis | ACTA1—pro-fibrotic factor |
| Increased Hypercytokinemia | ALCAM—increased cytokine production FHL1—increased cytokine translation |
| Increased Viral Transcription and Translation | CA12—translational control SYNE2—contributes to cell cycle regulation |
| Multi-Organ Failure | PCDH9—cadherin disruption decreases endothelial barrier integrity |
| Organ Specific Damage | HSPA1A—ischemia and kidney damage PDE3B—induces angiogenesis, contributes to ARDS SGCD—related to cardiomyopathy SLC39A14—related to ferroptosis in liver |
| Gene | Basic Functionality | Drug Target Evidence |
|---|---|---|
| ANPEP | Peptidase with increased activity in coronaviral infections | Unverified |
| CCL2 | Inflammatory cytokine upregulated in many viral infections | Unverified |
| HBB | Hemoglobin beta subunit that regulates inflammatory cytokines | Unverified |
| IFI27 | Interferon-induced protein upregulated in severe COVID-19 | Unverified |
| HLA-A | MHC Class I peptide upregulated in COVID-19 and liver injury | Unverified |
| HLA-B | MHC Class I peptide upregulated in COVID-19 and liver injury | Unverified |
| HLA-C | MHC Class I peptide upregulated in COVID-19 and liver injury | Unverified |
| IL-17A | Cytokine that is upregulated in severely infected patients | Use in other diseases |
| IL4R | Cytokine receptor that is upregulated in pulmonary disease | Use in other diseases |
| ALPI | Phosphatase that is upregulated in respiratory disease | Use in other diseases |
| MAPK8 | Kinase involved in apoptosis in SARS infections | Use in other diseases |
| SERPINC1 | Upregulated activity in cirrhosis of liver and COVID-19 | Validated COVID-19 Use |
| CCL7 | Inflammatory cytokine | Validated COVID-19 Use |
| AGTR1 | Increased activity appears in cirrhosis and SARS infections | Validated COVID-19 Use |
| TUBG2 | Structural molecule upregulated in viral infections | Validated COVID-19 Use |
| PPARG | Receptor upregulated in kidney injury and SARS infections | Validated COVID-19 Use |
| IL-6 | Pro-fibrotic factor and inflammatory cytokine | Validated COVID-19 Use |
| GABRA4 | GABA receptor with increased expression in SARS infections | Validated COVID-19 Use |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bass, A.; Liu, Y.; Dakshanamurthy, S. Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage. Viruses 2021, 13, 2418. https://doi.org/10.3390/v13122418
Bass A, Liu Y, Dakshanamurthy S. Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage. Viruses. 2021; 13(12):2418. https://doi.org/10.3390/v13122418
Chicago/Turabian StyleBass, Alexandrea, Yiran Liu, and Sivanesan Dakshanamurthy. 2021. "Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage" Viruses 13, no. 12: 2418. https://doi.org/10.3390/v13122418
APA StyleBass, A., Liu, Y., & Dakshanamurthy, S. (2021). Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage. Viruses, 13(12), 2418. https://doi.org/10.3390/v13122418