
Sendai Virus and a Unified Model of Mononegavirus RNA Synthesis

 , and
, and 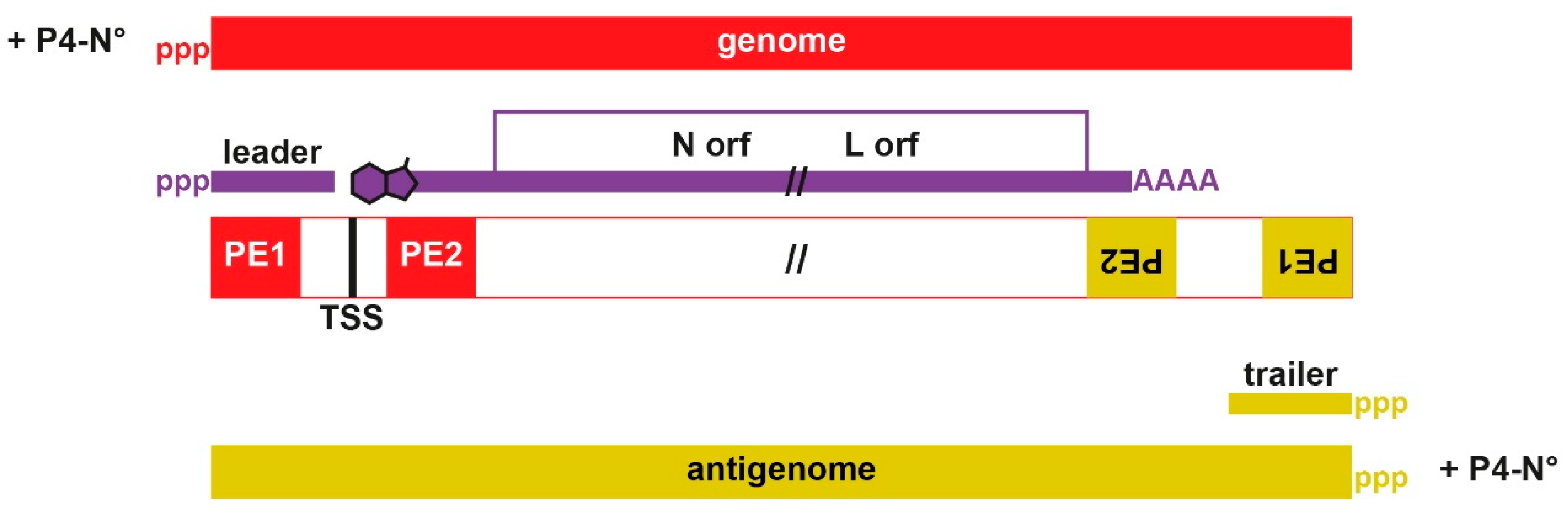{kind=link}
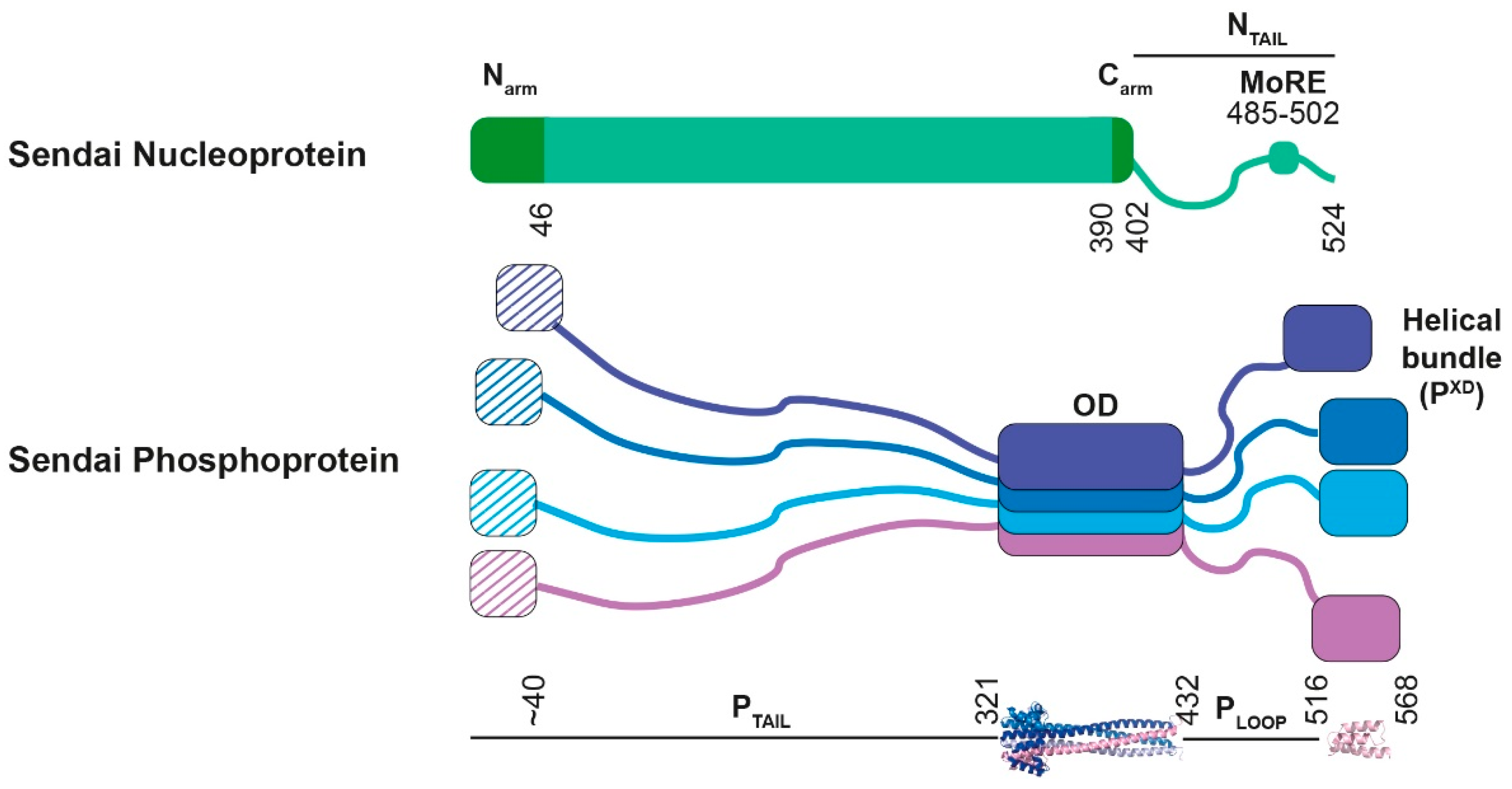{kind=link}
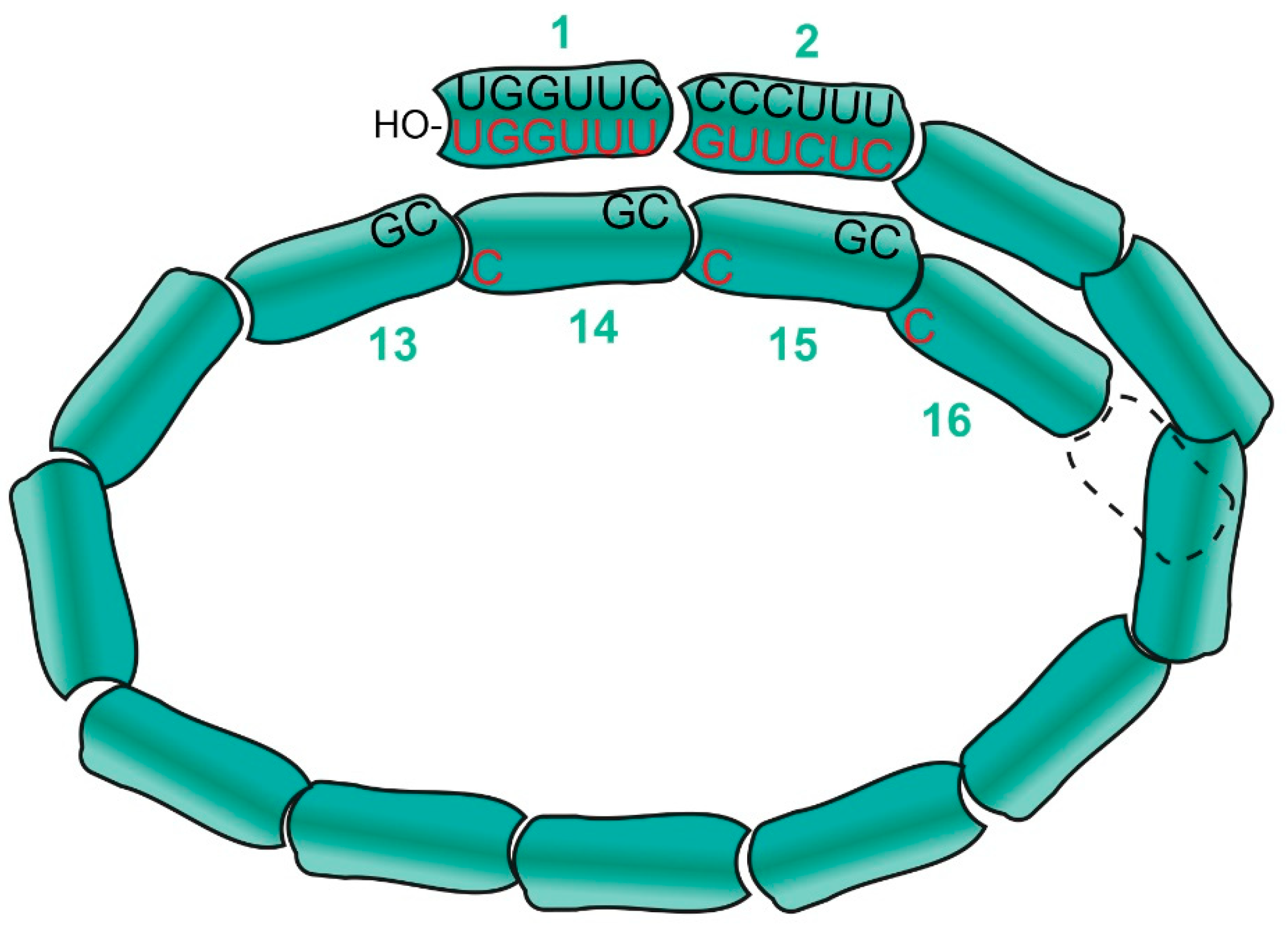{kind=link}
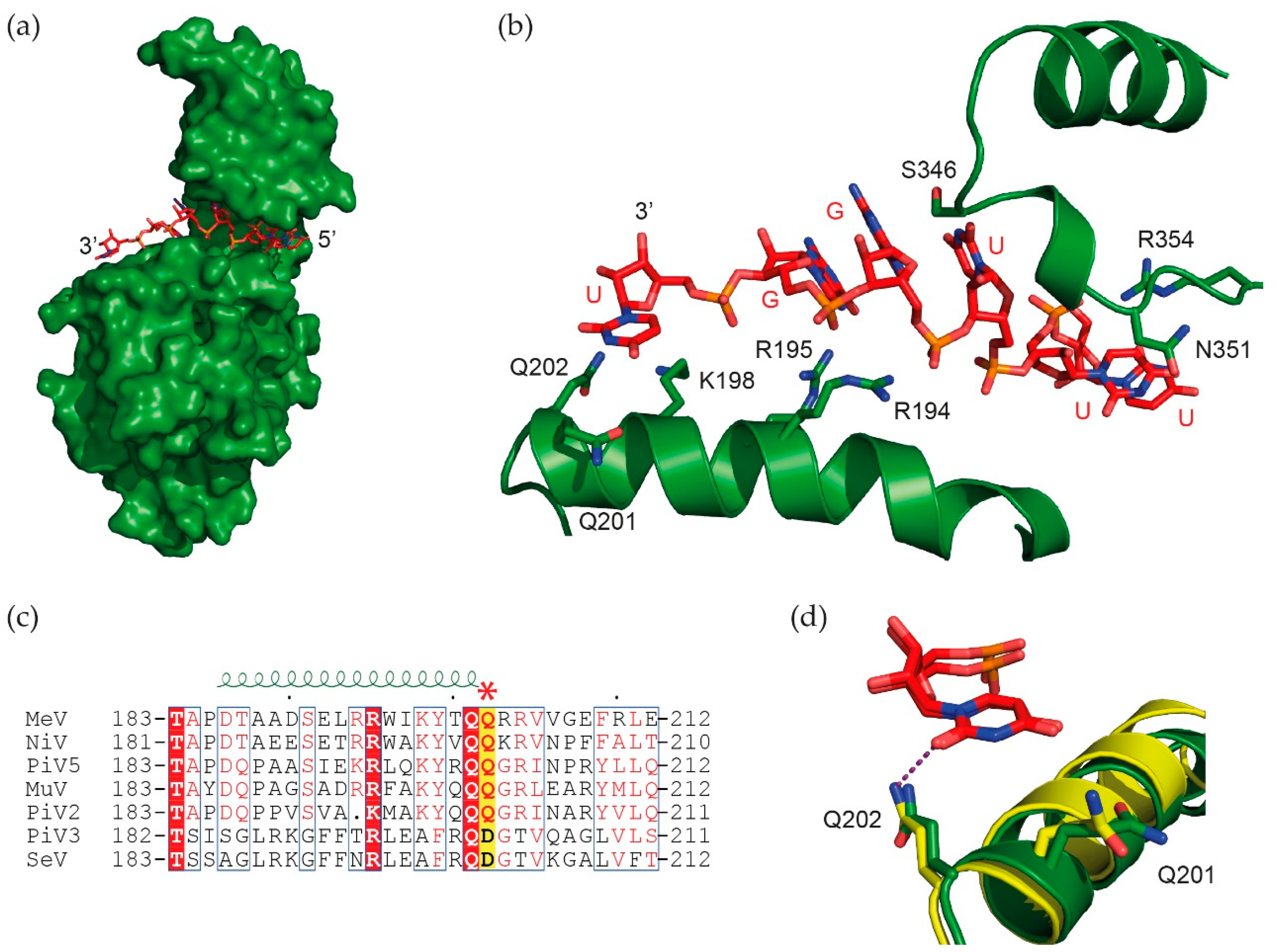{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mononegaviruses
2.1. MNV Nucleocapsids
2.2. MNV RNA Synthesis
2.3. 3′ end Promoters
2.4. Leader Regions and the Control of RNA Synthesis
2.5. The RdRp Template Channel and Recognition of Cis-Acting Signals

2.6. Coupling of Genome Synthesis and Nascent Chain Assembly during Replication
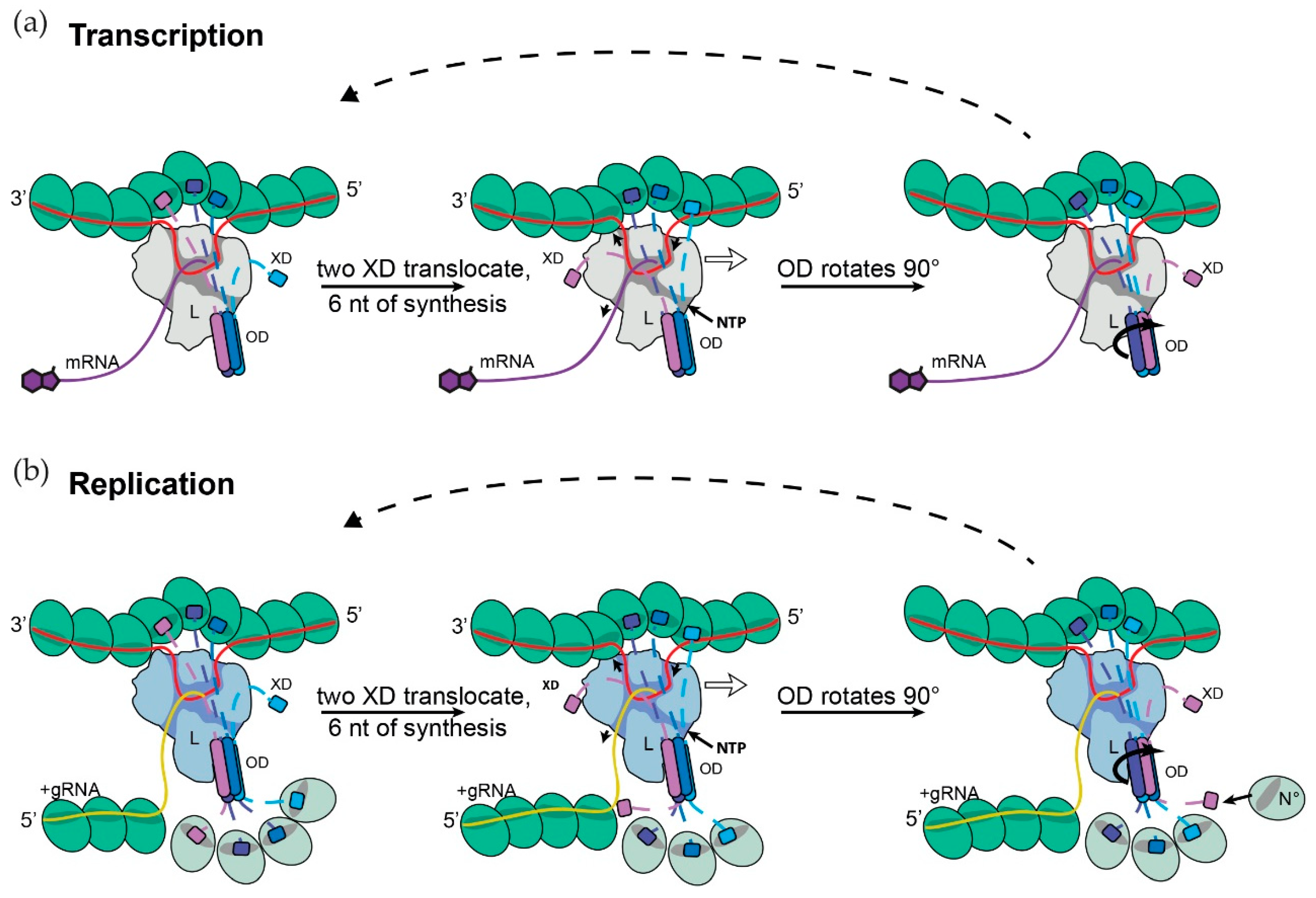
2.7. Model
3. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Alayyoubi, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc. Natl. Acad. Sci. USA 2015, 112, E1792–E1799. [Google Scholar] [CrossRef] [Green Version]
- Albertini, A.A.V.; Wernimont, A.K.; Muziol, T.M.; Ravelli, R.B.G.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W.H. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 2006, 313, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Zhang, X.; Wertz, G.W.; Luo, M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 2006, 313, 357–360. [Google Scholar] [CrossRef]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagné, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Shan, H.; Liu, M.; Li, T.; Luo, R.; Yang, L.; Qi, L.; Chu, X.; Su, X.; Wang, R.; et al. Structure and assembly of double-headed Sendai virus nucleocapsids. Commun. Biol. 2021, 4, 494. [Google Scholar] [CrossRef]
- Lynch, S.; Kolakofsky, D. Ends of the RNA within Sendai virus defective interfering nucleocapsids are not free. J. Virol. 1978, 28, 584–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, J.; Pelet, T.; Kolakofsky, D. An acidic activation-like domain of the Sendai virus P protein is required for RNA synthesis and encapsidation. Virology 1994, 202, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Jensen, M.R.; Communie, G.; Maurin, D.; Schoehn, G.; Ruigrok, R.W.H.; Blackledge, M. Self-assembly of measles virus nucleocapsid-like particles: Kinetics and RNA sequence dependence. Angew. Chem. Int. Ed. Engl. 2016, 55, 9356–9360. [Google Scholar] [CrossRef] [Green Version]
- Gubbay, O.; Curran, J.; Kolakofsky, D. Sendai virus genome synthesis and assembly are coupled: A possible mechanism to promote viral RNA polymerase processivity. J. Gen. Virol. 2001, 82, 2895–2903. [Google Scholar] [CrossRef]
- Iverson, L.E.; Rose, J.K. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell 1981, 23, 477–484. [Google Scholar] [CrossRef]
- Plumet, S.; Duprex, W.P.; Gerlier, D. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 2005, 79, 6900–6908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, J.; Marq, J.B.; Kolakofsky, D. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 1995, 69, 849–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarbouriech, N.; Curran, J.; Ruigrok, R.W.H.; Burmeister, W.P. Tetrameric coiled coil domain of Sendai virus phospho-protein. Nat. Struct. Biol. 2000, 7, 777–781. [Google Scholar] [PubMed]
- Guseva, S.; Milles, S.; Jensen, M.R.; Salvi, N.; Kleman, J.-P.; Maurin, D.; Ruigrok, R.W.H.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [Green Version]
- Milles, S.; Jensen, M.R.; Lazert, C.; Guseva, S.; Ivashchenko, S.; Communie, G.; Maurin, D.; Gerlier, D.; Ruigrok, R.W.H.; Blackledge, M. An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 2018, 4, eaat7778. [Google Scholar] [CrossRef] [Green Version]
- Bloyet, L.-M.; Brunel, J.; Dosnon, M.; Hamon, V.; Erales, J.; Gruet, A.; Lazert, C.; Bignon, C.; Roche, P.; Longhi, S.; et al. Modulation of re-initiation of measles virus transcription at intergenic regions by PXD to NTAIL binding strength. PLoS Pathog. 2016, 12, e1006058. [Google Scholar] [CrossRef] [Green Version]
- Krumm, S.A.; Takeda, M.; Plemper, R.K. The measles virus nucleocapsid protein tail domain is dispensable for viral polymerase recruitment and activity. J. Biol. Chem. 2013, 288, 29943–29953. [Google Scholar] [CrossRef] [Green Version]
- Sourimant, J.; Thakkar, V.D.; Cox, R.M.; Plemper, R.K. Viral evolution identifies a regulatory interface between paramyxovirus polymerase complex and nucleocapsid that controls replication dynamics. Sci. Adv. 2020, 6, eaaz1590. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.K.; Ito, Y.; Parks, G.D. A functional antigenomic promoter for the paramyxovirus simian virus 5 requires proper spacing between an essential internal segment and the 3’ terminus. J. Virol. 1998, 72, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.K.; Parks, G.D. RNA replication for the paramyxovirus simian virus 5 requires an internal repeated (CGNNNN) sequence motif. J. Virol. 1999, 73, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Tapparel, C.; Maurice, D.; Roux, L. The activity of Sendai virus genomic and antigenomic promoters requires a second element past the leader template regions: A motif (GNNNNN)3 is essential for replication. J. Virol. 1998, 72, 3117–3128. [Google Scholar] [CrossRef] [Green Version]
- Weik, M.; Enterlein, S.; Schlenz, K.; Mühlberger, E. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J. Virol. 2005, 79, 10660–10671. [Google Scholar] [CrossRef] [Green Version]
- Calain, P.; Roux, L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J. Virol. 1993, 67, 4822–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolakofsky, D. Paramyxovirus RNA synthesis, mRNA editing, and genome hexamer phase: A review. Virology 2016, 498, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Desfosses, A.; Milles, S.; Jensen, M.R.; Guseva, S.; Colletier, J.-P.; Maurin, D.; Schoehn, G.; Gutsche, I.; Ruigrok, R.W.H.; Blackledge, M. Assembly and cryo-EM structures of RNA-specific measles virus nucleocapsids provide mechanistic insight into paramyxoviral replication. Proc. Natl. Acad. Sci. USA 2019, 116, 4256–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoehn, G.; Mavrakis, M.; Albertini, A.; Wade, R.; Hoenger, A.; Ruigrok, R.W.H. The 12 Å structure of trypsin-treated measles virus N-RNA. J. Mol. Biol. 2004, 339, 301–312. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Ohta, K.; Kolakofsky, D.; Nishio, M. A point mutation in the RNA-binding domain of human parainfluenza virus type 2 nucleoprotein elicits abnormally enhanced polymerase activity. J. Virol. 2017, 91, e02203-16. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ohta, K.; Kolakofsky, D.; Nishio, M. The control of paramyxovirus genome hexamer length and mRNA editing. RNA 2018, 24, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Iseni, F.; Baudin, F.; Garcin, D.; Marq, J.-B.; Ruigrok, R.W.H.; Kolakofsky, D. Chemical modification of nucleotide bases and mRNA editing depend on hexamer or nucleoprotein phase in Sendai virus nucleocapsids. RNA 2002, 8, 1056–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrodinger, LLC, The PyMOL Molecular Graphics System, Version 1.8. In 2015. Available online: https://pymol.org/2/support.html (accessed on 9 November 2021).
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Le Mercier, P.; Kolakofsky, D. Bipartite promoters and RNA editing of paramyxoviruses and filoviruses. RNA 2019, 25, 279–285. [Google Scholar] [CrossRef]
- Bach, S.; Biedenkopf, N.; Gruenweller, A.; Becker, S.; Hartmann, R.K. Hexamer phasing governs transcription initiation in the 3′-leader of Ebola virus. RNA 2020, 26, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Shabman, R.; Jabado, O.J.; Mire, C.E.; Stockwell, T.; Edwards, M.; Mahajan, M.; Geisbert, T.W.; Basler, C.F. Deep sequencing identifies noncanonical editing of Ebola and Marburg virus RNAs in infected cells. mBio 2014, 5, e02011-14. [Google Scholar] [CrossRef] [Green Version]
- Colonno, R.J.; Banerjee, A.K. In vitro RNA transcription by the New Jersey serotype of vesicular stomatitis virus. II. Characterization of the leader RNA. J. Virol. 1978, 26, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppert, M.; Rittenhouse, L.; Perrault, J.; Summers, D.F.; Kolakofsky, D. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell 1979, 18, 735–747. [Google Scholar] [CrossRef]
- Blumberg, B.M.; Giorgi, C.; Kolakofsky, D. N protein of vesicular stomatitis virus selectively encapsidates leader RNA in vitro. Cell 1983, 32, 559–567. [Google Scholar] [CrossRef]
- Iseni, F.; Baudin, F.; Blondel, D.; Ruigrok, R.W.H. Structure of the RNA inside the vesicular stomatitis virus nucleocapsid. RNA 2000, 6, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Iseni, F.; Garcin, D.; Nishio, M.; Kedersha, N.; Anderson, P.; Kolakofsky, D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J. 2002, 21, 5141–5150. [Google Scholar] [CrossRef] [Green Version]
- Hinzman, E.E.; Barr, J.N.; Wertz, G.W. Identification of an upstream sequence element required for vesicular stomatitis virus mRNA transcription. J. Virol. 2002, 76, 7632–7641. [Google Scholar] [CrossRef] [Green Version]
- Plattet, P.; Strahle, L.; Le Mercier, P.; Hausmann, S.; Garcin, D.; Kolakofsky, D. Sendai virus RNA polymerase scanning for mRNA start sites at gene junctions. Virology 2007, 362, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Li, Z.; Jenni, S.; Rahmeh, A.A.; Morin, B.M.; Grant, T.; Grigorieff, N.; Harrison, S.C.; Whelan, S.P.J. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 2015, 162, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, X.; Lattmann, S.; El Sahili, A.; Yeo, T.H.; Jia, H.; Cressey, T.; Ludeke, B.; Noton, S.; Kalocsay, M.; et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nat. Cell Biol. 2020, 577, 275–279. [Google Scholar] [CrossRef]
- Xie, J.; Wang, L.; Zhai, G.; Wu, D.; Lin, Z.; Wang, M.; Yan, X.; Gao, L.; Huang, X.; Fearns, R.; et al. Structural architecture of a dimeric paramyxovirus polymerase complex. bioRxiv 2001. preprint. [Google Scholar] [CrossRef]
- Cao, D.; Gao, Y.; Roesler, C.; Rice, S.; D’Cunha, P.; Zhuang, L.; Slack, J.; Domke, M.; Antonova, A.; Romanelli, S.; et al. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat. Commun. 2020, 11, 368. [Google Scholar] [CrossRef] [Green Version]
- Gilman, M.S.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eléouët, J.-F.; et al. Structure of the respiratory syncytial virus polymerase complex. Cell 2019, 179, 193–204.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reguera, J.; Gerlach, P.; Cusack, S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr. Opin. Struct. Biol. 2016, 36, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Curran, J. Reexamination of the Sendai virus P protein domains required for RNA synthesis: A possible supplemental role for the P protein. Virology 1996, 221, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Wandzik, J.M.; Kouba, T.; Karuppasamy, M.; Pflug, A.; Drncova, P.; Provaznik, J.; Azevedo, N.; Cusack, S. A structure-based model for the complete transcription cycle of influenza polymerase. Cell 2020, 181, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, S.; Garcin, D.; Delenda, C.; Kolakofsky, D. The versatility of paramyxovirus RNA polymerase stuttering. J. Virol. 1999, 73, 5568–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, S.; Garcin, D.; Morel, A.S.; Kolakofsky, D. Two nucleotides immediately upstream of the essential A6G3 slippery sequence modulate the pattern of G insertions during Sendai virus mRNA editing. J. Virol. 1999, 73, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Arnheiter, H.; Davis, N.L.; Wertz, G.; Schubert, M.; Lazzarini, R.A. Role of the nucleocapsid protein in regulating vesicular stomatitis virus RNA synthesis. Cell 1985, 41, 259–267. [Google Scholar] [CrossRef]
- Vidal, S.; Kolakofsky, D. Modified model for the switch from Sendai virus transcription to replication. J. Virol. 1989, 63, 1951–1958. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein in complex with an amino-terminal peptide of the phosphoprotein. J. Virol. 2018, 92, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.-M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.H.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles virus forms inclusion bodies with properties of liquid organelles. J. Virol. 2019, 93, e00948-19. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Green, T.J.; Purushotham, S.; Deivanayagam, C.; Bedwell, G.J.; Prevelige, P.E.; Luo, M. Structural and functional characterization of the mumps virus phosphoprotein. J. Virol. 2013, 87, 7558–7568. [Google Scholar] [CrossRef] [Green Version]
- Nilsson-Payant, B.E.; Blanco-Melo, D.; Uhl, S.; Escudero-Perez, B.; Olschewski, S.; Thibault, P.; Panis, M.; Rosenthal, M.; Munoz-Fontela, C.; Lee, B.; et al. Reduced nucleoprotein availability impairs negative-dense RNA virus rep-lication and promotes host recognition. J. Virol. 2021, 95, e02274-20. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolakofsky, D.; Le Mercier, P.; Nishio, M.; Blackledge, M.; Crépin, T.; Ruigrok, R.W.H. Sendai Virus and a Unified Model of Mononegavirus RNA Synthesis. Viruses 2021, 13, 2466. https://doi.org/10.3390/v13122466
Kolakofsky D, Le Mercier P, Nishio M, Blackledge M, Crépin T, Ruigrok RWH. Sendai Virus and a Unified Model of Mononegavirus RNA Synthesis. Viruses. 2021; 13(12):2466. https://doi.org/10.3390/v13122466
Chicago/Turabian StyleKolakofsky, Daniel, Philippe Le Mercier, Machiko Nishio, Martin Blackledge, Thibaut Crépin, and Rob W. H. Ruigrok. 2021. "Sendai Virus and a Unified Model of Mononegavirus RNA Synthesis" Viruses 13, no. 12: 2466. https://doi.org/10.3390/v13122466
APA StyleKolakofsky, D., Le Mercier, P., Nishio, M., Blackledge, M., Crépin, T., & Ruigrok, R. W. H. (2021). Sendai Virus and a Unified Model of Mononegavirus RNA Synthesis. Viruses, 13(12), 2466. https://doi.org/10.3390/v13122466