
Human Respiratory Syncytial Virus Infection in a Human T Cell Line Is Hampered at Multiple Steps

 , ,
, ,  and
and 
Abstract
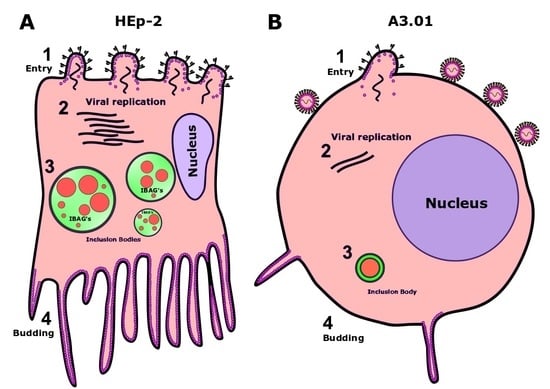
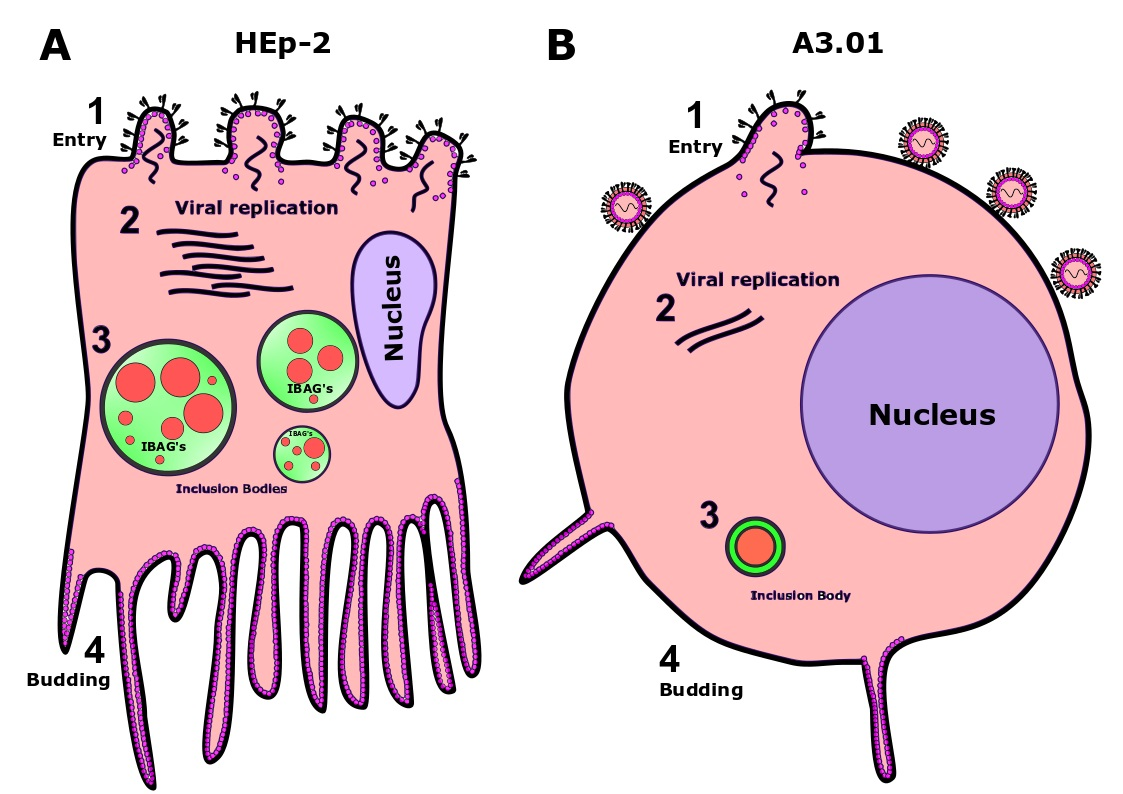
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies
2.3. Flow Cytometry
2.4. Biobond and Poly-Lysine Coverslips Treatment
2.5. Immunofluorescence
2.6. Fluorescent In Situ Hybridization
2.7. Quantitative Real-Time RT-PCR (RT-qPCR)
2.8. R18 Fluorescence Conjugation and Dequenching Assay
2.9. Fluorescent Focus Assay
2.10. Immunoblotting
2.11. Statistical Analysis
3. Results
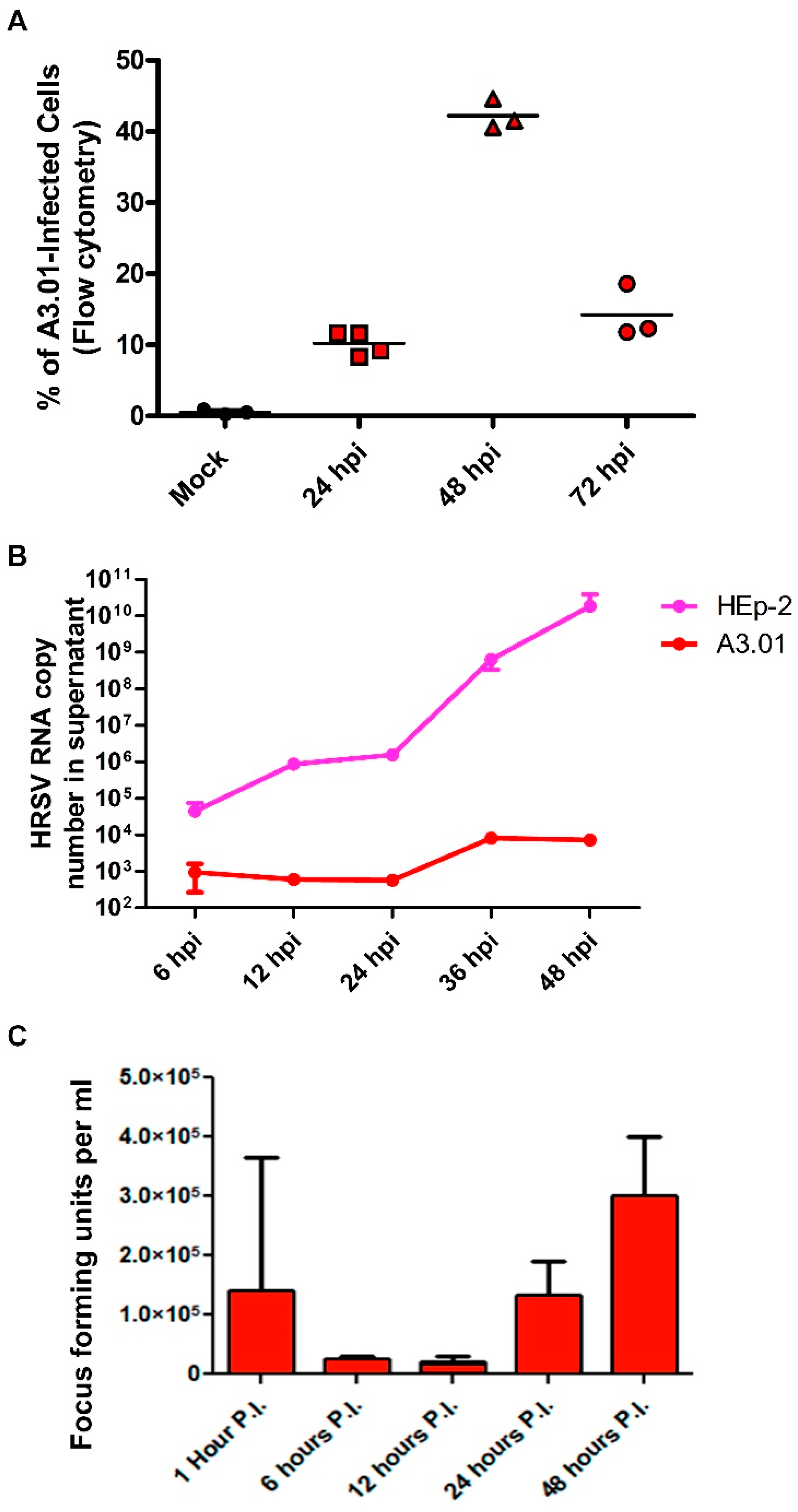
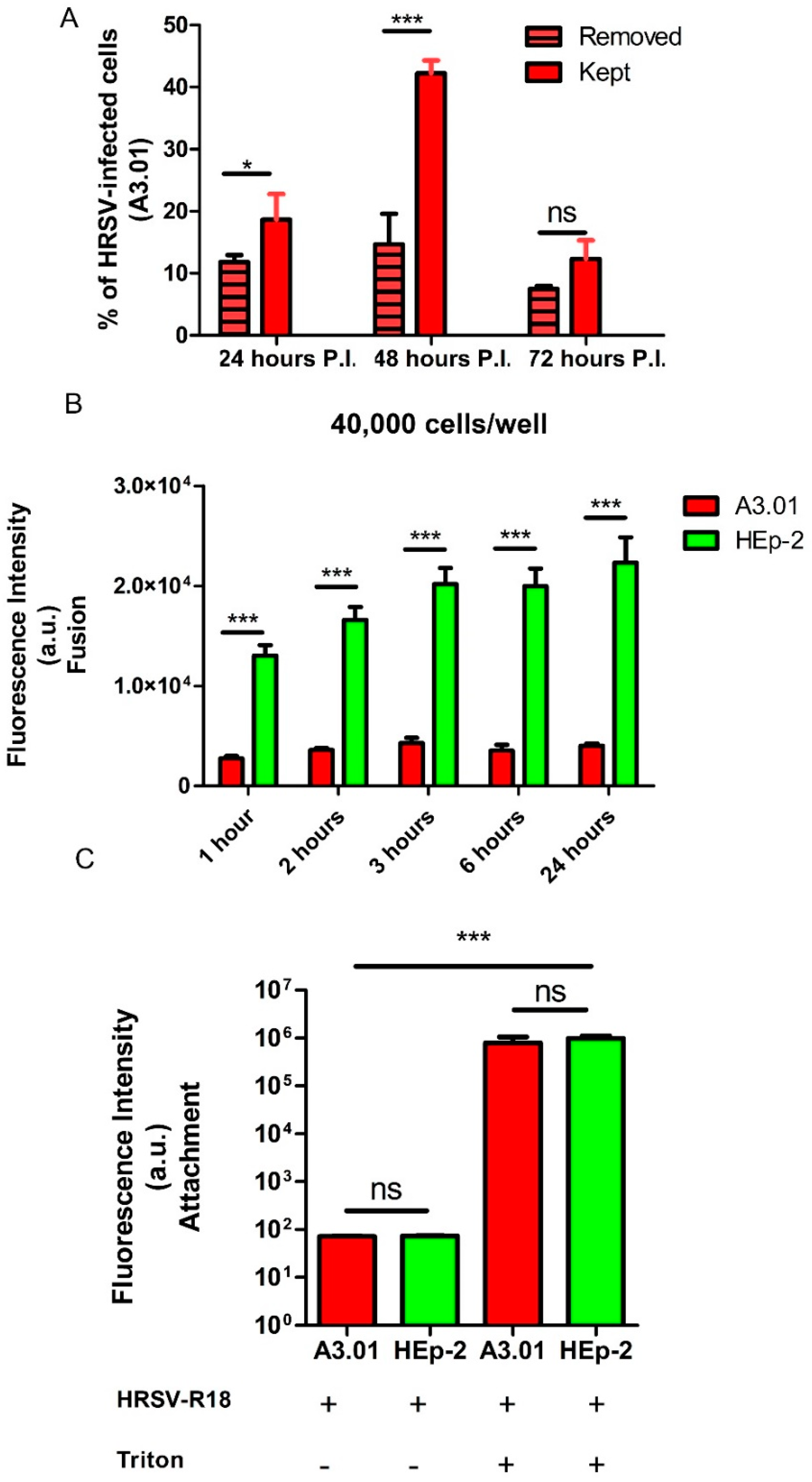
3.1. A3.01 Lymphocytes Inoculated with HRSV Are Inefficient in Progeny Production
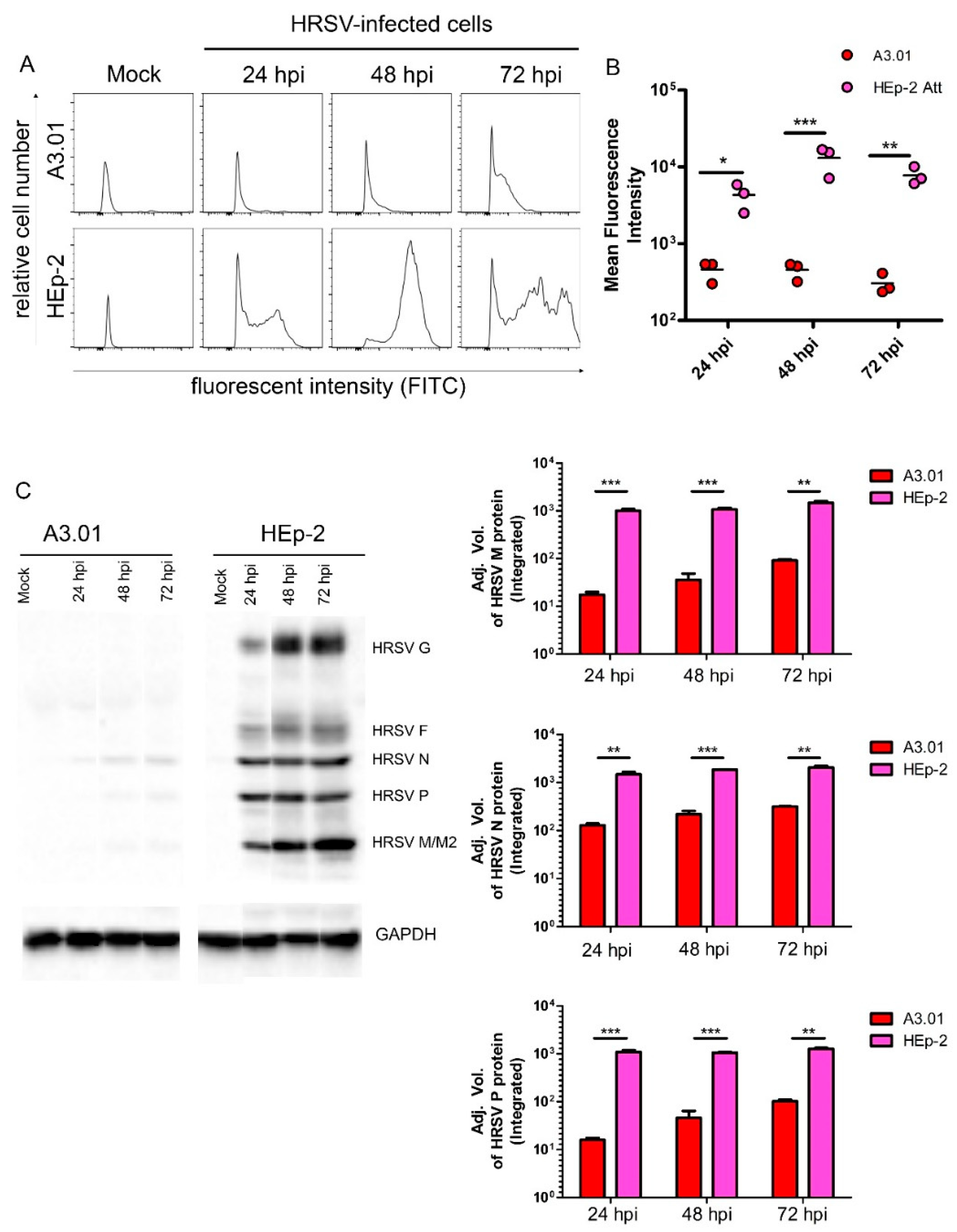
3.2. HRSV Genome Replication in A3.01 Cells Is Inefficient
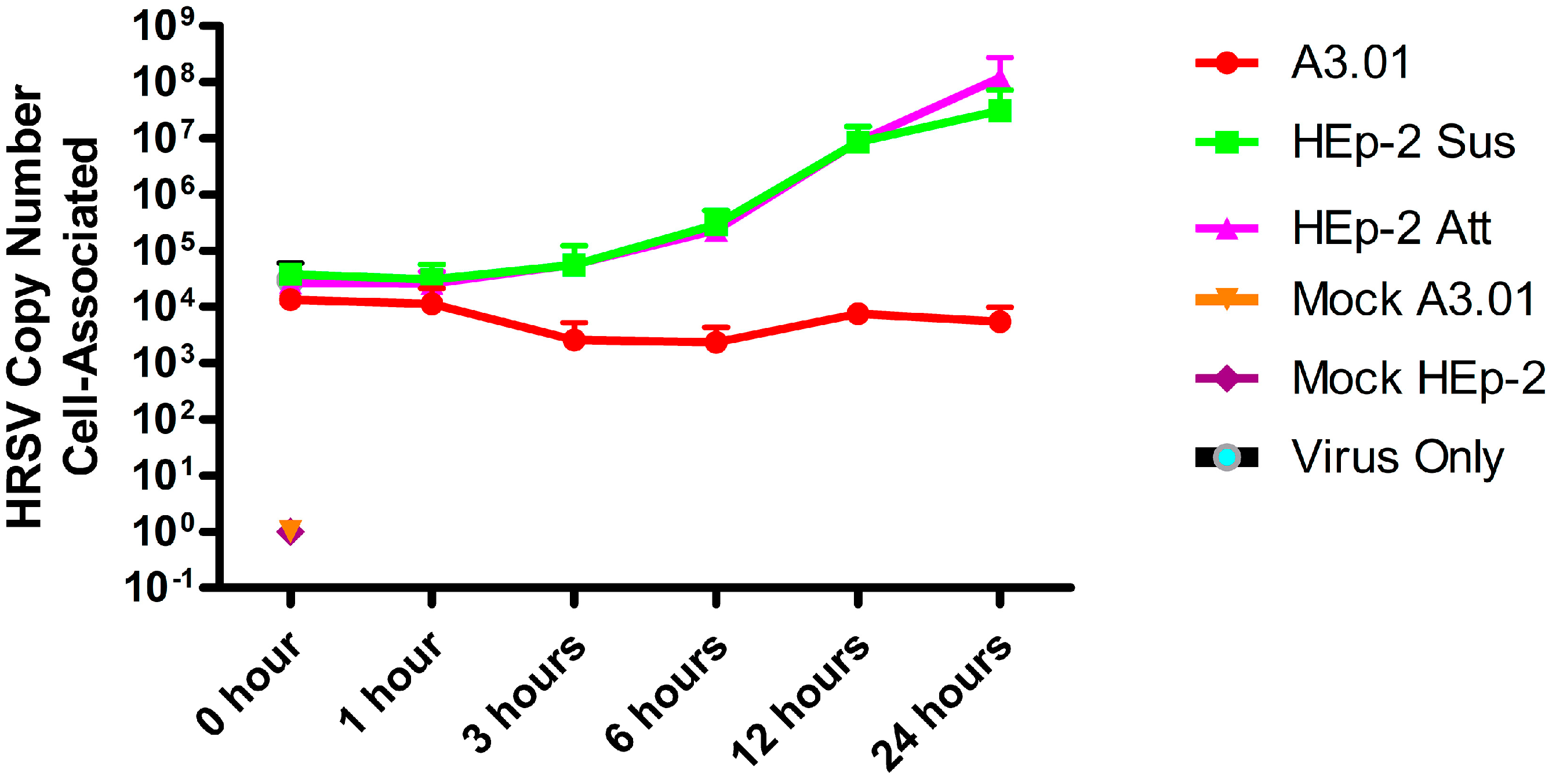
3.3. The Fusion Process of HRSV in A3.01 Cells Is Defective
3.4. HRSV Protein Production in A3.01 Cells Is Small
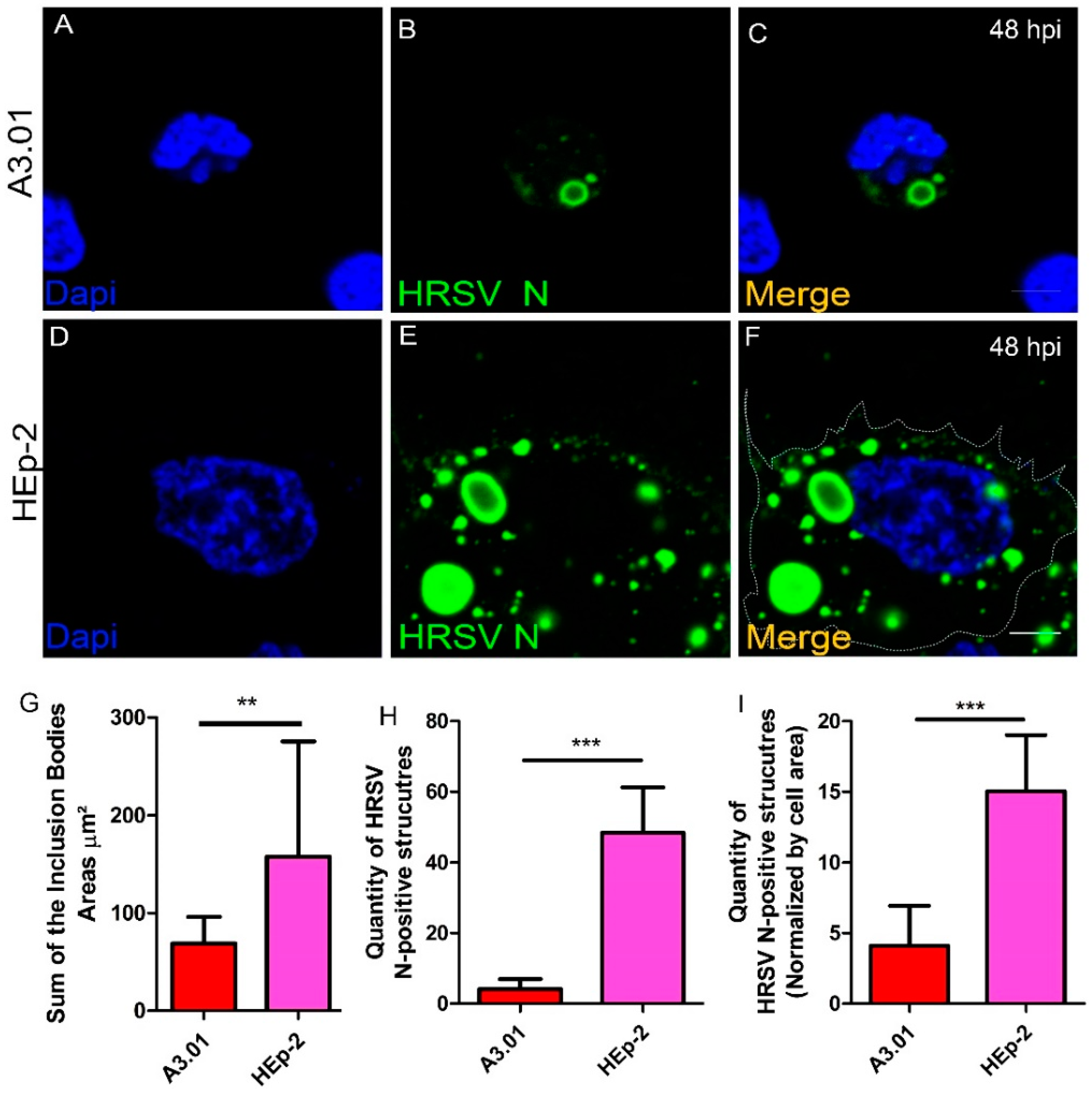
3.5. HRSV Inclusion Body Formation is Compromised in A3.01
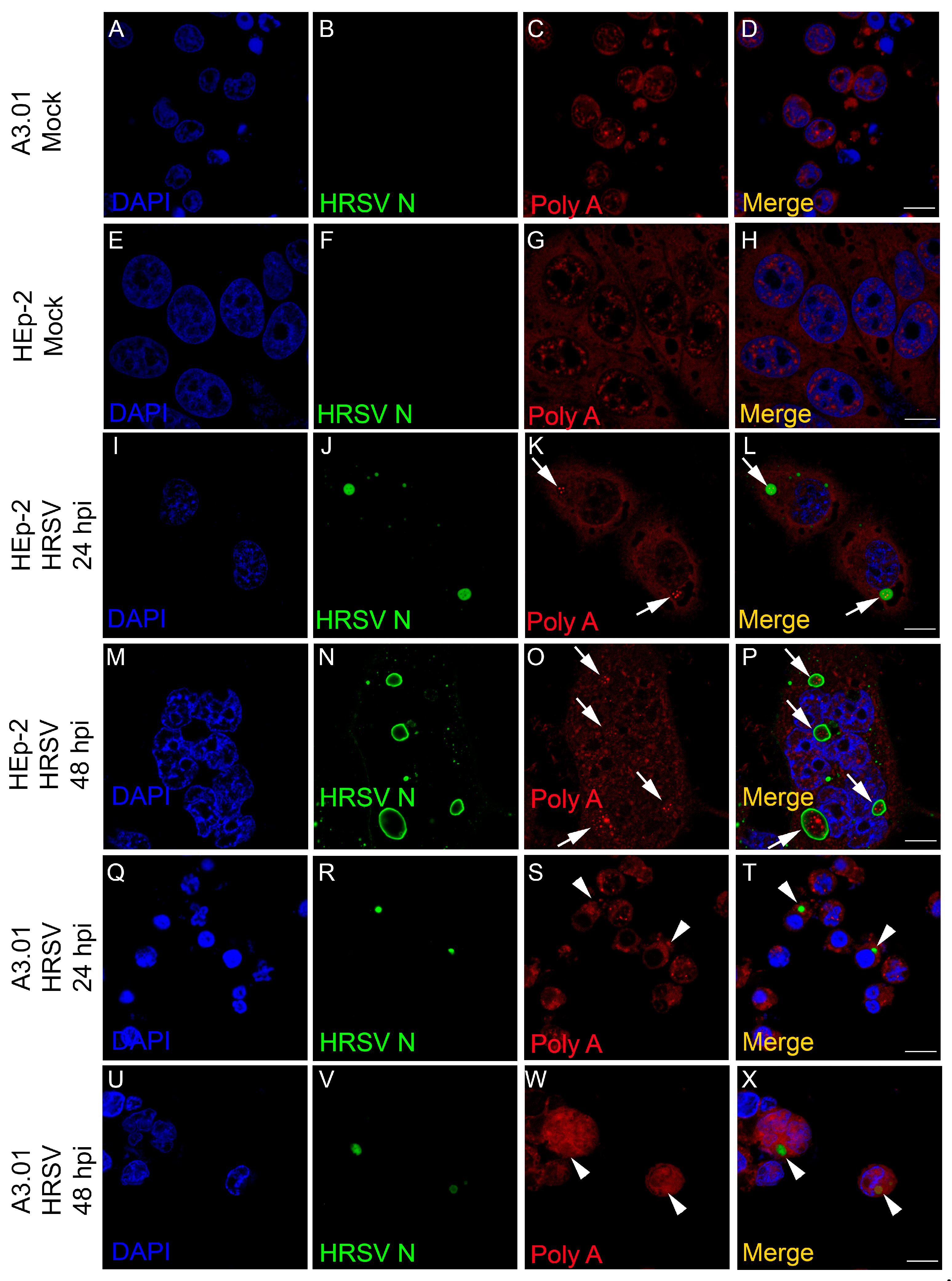
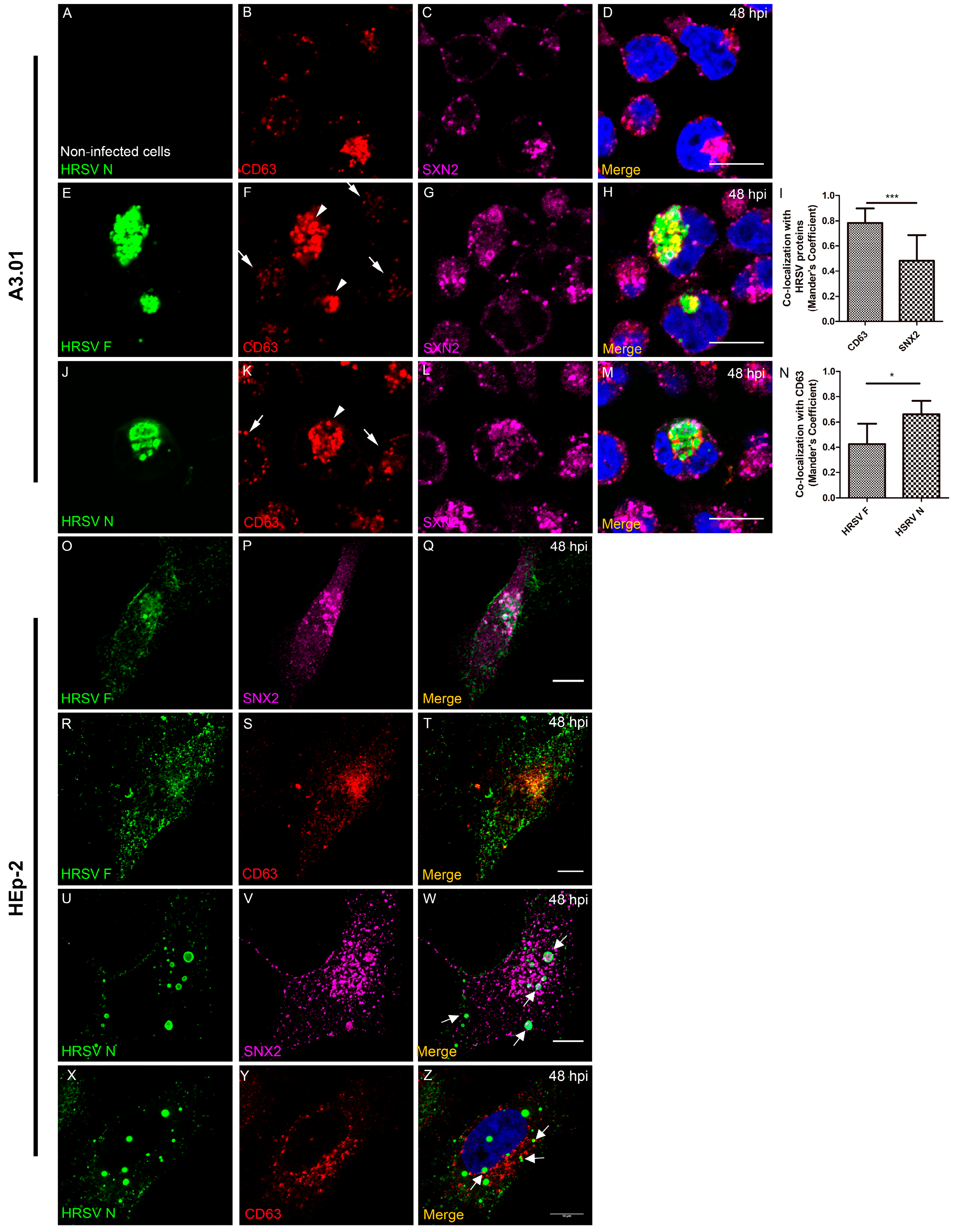
3.6. The Inclusion Bodies of HRSV in A3.01 Cells Lack IBAGs
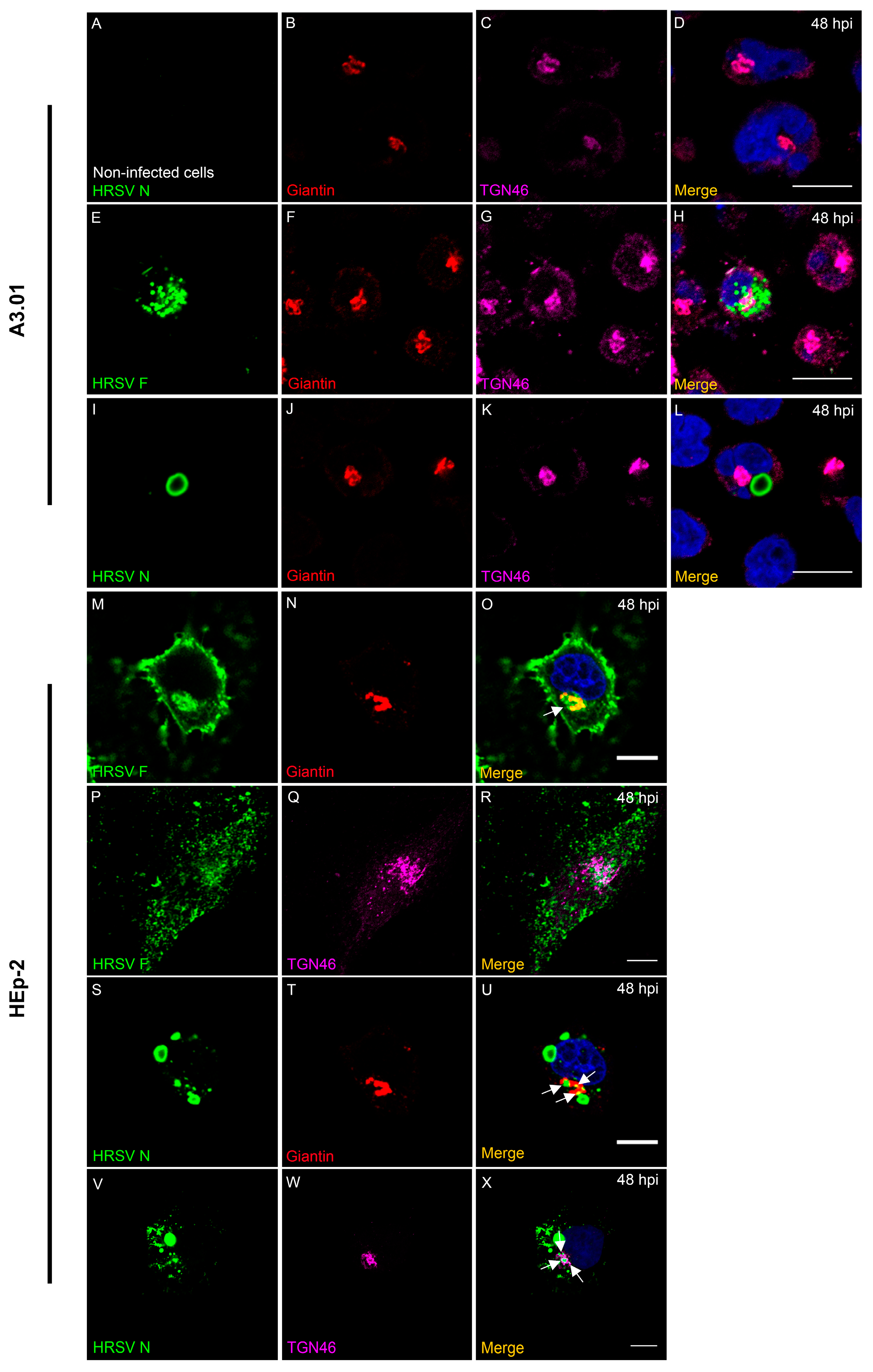
3.7. HRSV F Protein Partially Colocalizes with Golgi Markers Giantin and TGN46 in A3.01 Cells
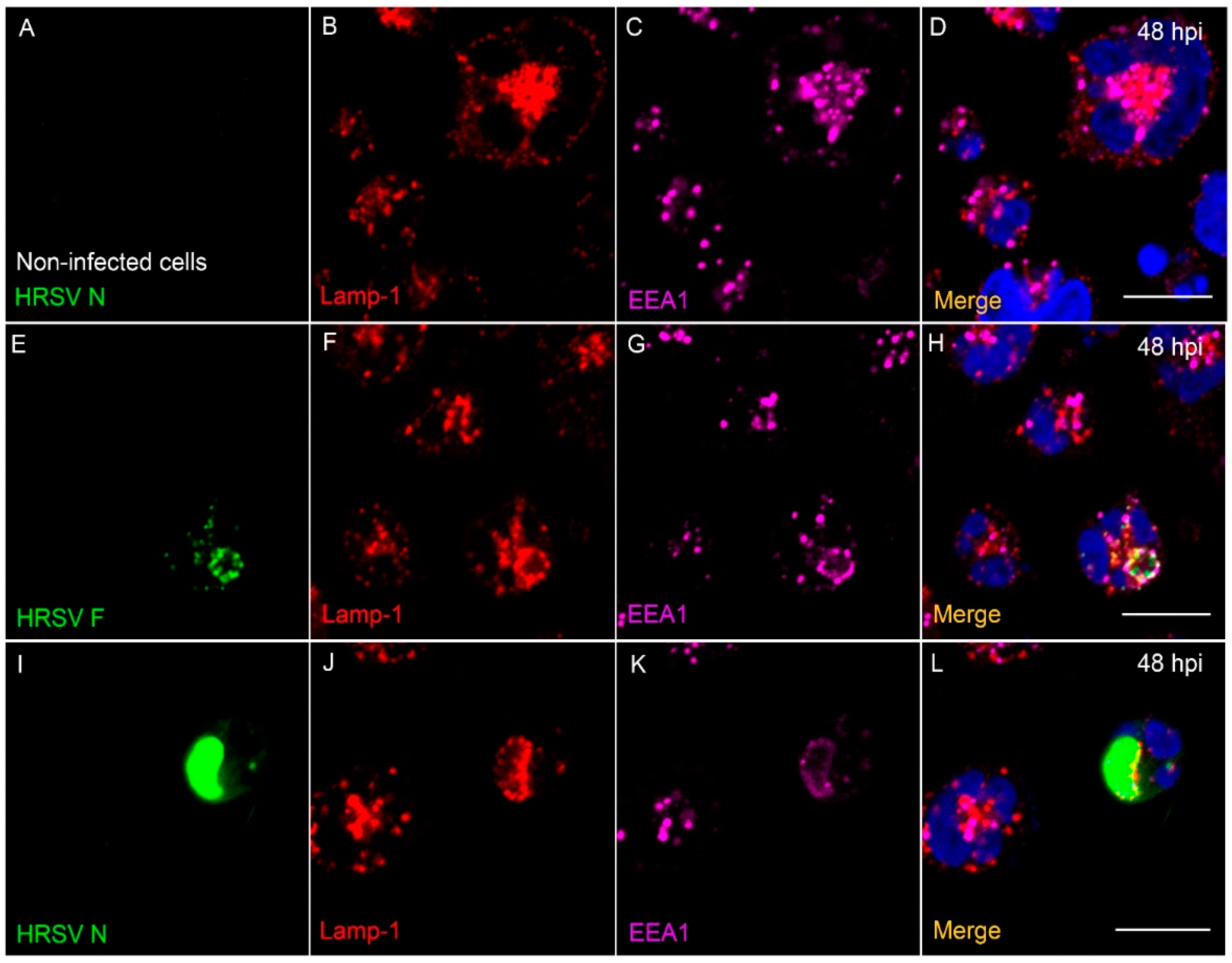
3.8. The HRSV F and N Proteins Partially Co-Localize with Markers of Endosomal Pathway
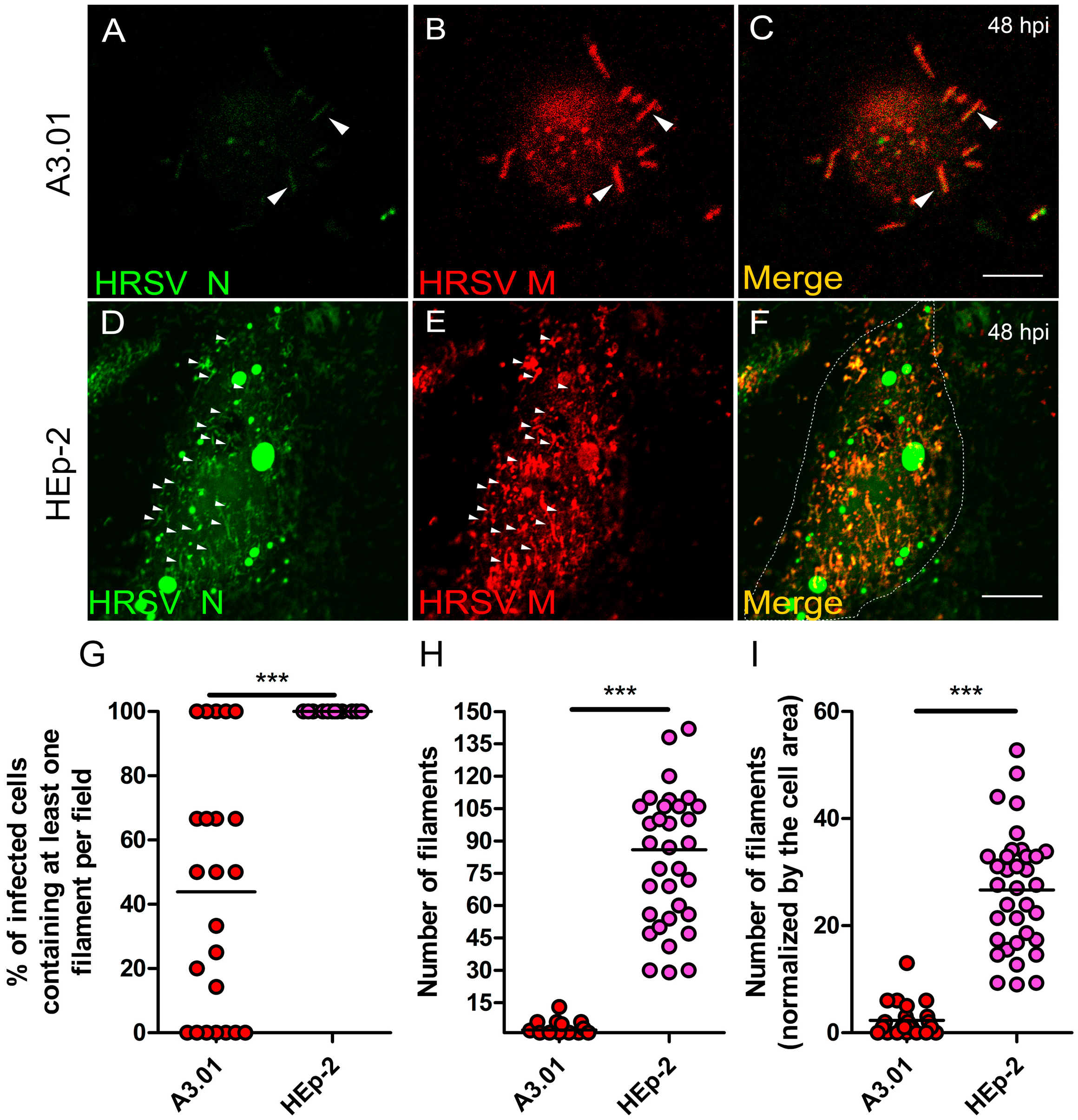
3.9. The Production of HRSV Filaments at the Plasma Membrane of A3.01 Cells Is Very Low
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, P.L.; Karron, R.A. Respiratory Syncytial Virus and Metapneumovirus. In Fields Virology, 6th ed.; Knipe, M.D., Howley, M.P., Eds.; Wolters Kluwer, Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1086–1123. [Google Scholar]
- Rezaee, F.; Gibson, L.F.; Piktel, D.; Othumpangat, S.; Piedimonte, G. Respiratory syncytial virus infection in human bone marrow stromal cells. Am. J. Respir Cell Mol. Biol. 2011, 45, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Mills, B.G.; Singer, F.R.; Weiner, L.P.; Holst, P.A. Immunohistological demonstration of respiratory syncytial virus antigens in Paget disease of bone. Proc. Natl. Acad. Sci. USA 1981, 78, 1209–1213. [Google Scholar] [CrossRef] [Green Version]
- Modena, J.L.; Valera, F.C.; Jacob, M.G.; Buzatto, G.P.; Saturno, T.H.; Lopes, L.; Souza, J.M.; Paula, F.E.; Silva, M.L.; Carenzi, L.R.; et al. High rates of detection of respiratory viruses in tonsillar tissues from children with chronic adenotonsillar disease. PLoS ONE 2012, 7, e42136. [Google Scholar] [CrossRef]
- Raiden, S.; Sananez, I.; Remes-Lenicov, F.; Pandolfi, J.; Romero, C.; De Lillo, L.; Ceballos, A.; Geffner, J.; Arruvito, L. Respiratory syncytial virus (RSV) infects CD4+ T cells: Frequency of circulating CD4+ RSV+ T cells as a marker of disease severity in young children. J. Infect. Dis. 2017, 215, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Levine, S.; Kaliaber-Franco, R.; Paradiso, P.R. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J. Gen. Virol. 1987, 68, 2521–2524. [Google Scholar] [CrossRef]
- Feldman, S.A.; Audet, S.; Beeler, J.A. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 2000, 74, 6442–6447, Updated. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.M.; Young, T.F.; Tucker, S.P.; Mackay, J.P. The core of the respiratory syncytial virus fusion protein is a trimeric coiled coil. J. Virol. 2000, 74, 5911–5920. [Google Scholar] [CrossRef] [Green Version]
- Krzyzaniak, M.A.; Zumstein, M.T.; Gerez, J.A.; Picotti, P.; Helenius, A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog. 2013, 9, e1003309. [Google Scholar] [CrossRef] [Green Version]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef]
- García, J.; García-Barreno, B.; Vivo, A.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef]
- Carromeu, C.; Simabuco, F.M.; Tamura, R.E.; Farinha Arcieri, L.E.; Ventura, A.M. Intracellular localization of human respiratory syncytial virus L protein. Arch. Virol. 2007, 152, 2259–2263. [Google Scholar] [CrossRef]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [Green Version]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.-F.; Rameix-Welti, M.-A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Brock, S.C.; Heck, J.M.; McGraw, P.A.; Crowe, J.E. The transmembrane domain of the respiratory syncytial virus F protein is an orientation-independent apical plasma membrane sorting sequence. J. Virol. 2005, 79, 12528–12535. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.Y.; Utley, T.J.; Craven, R.E.; Rogers, M.C.; Lapierre, L.A.; Goldenring, J.R.; Crowe, J.E., Jr. Respiratory syncytial virus assembles into structured filamentous virion particles independently of host cytoskeleton and related proteins. PLoS ONE 2012, 7, e40826. [Google Scholar] [CrossRef]
- Anderson, K.; Stott, E.J.; Wertz, G.W. Intracellular processing of the human respiratory syncytial virus fusion glycoprotein: Amino acid substitutions affecting folding, transport and cleavage. J. Gen. Virol. 1992, 73, 1177–1188. [Google Scholar] [CrossRef]
- Kwilas, S.; Liesman, R.M.; Zhang, L.; Walsh, E.; Pickles, R.J.; Peeples, M.E. Respiratory Syncytial Virus Grown in Vero Cells Contains a Truncated Attachment Protein That Alters Its Infectivity and Dependence on Glycosaminoglycans. J. Virol. 2009, 83, 10710–10718. [Google Scholar] [CrossRef] [Green Version]
- Brock, S.C.; Goldenring, J.R.; Crowe, J.E. Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15143–15148. [Google Scholar] [CrossRef] [Green Version]
- Förster, A.; Maertens, G.N.; Farrell, P.J.; Bajorek, M. Dimerization of Matrix protein is required for budding of Respiratory Syncytial Virus. J. Virol. 2015, 89, JVI.03500-14. [Google Scholar] [CrossRef] [Green Version]
- Henderson, G.; Murray, J.; Yeo, R.P. Sorting of the Respiratory Syncytial Virus Matrix Protein into Detergent-Resistant Structures Is Dependent on Cell-Surface Expression of the Glycoproteins. Virology 2002, 300, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C.-A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M.-A.; Eléouët, J.-F. Identification and Characterization of the Binding Site of the Respiratory Syncytial Virus Phosphoprotein to RNA-Free Nucleoprotein. J. Virol. 2015, 89, 3484–3496. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, P.F.; Bueno, S.M.; Ramírez, B.A.; Gomez, R.S.; Riquelme, S.A.; Palavecino, C.E.; Mackern-Oberti, J.P.; Mora, J.E.; Depoil, D.; Sacristán, C.; et al. Surface expression of the hRSV nucleoprotein impairs immunological synapse formation with T cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3214–E3223. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, R.S.; Tavares, L.A.; Jesus, B.L.S.; Criado, M.F.; de Carvalho, A.N.; De Souza, J.P.; Bedi, S.; De Souza, M.M.; Silva, M.L.; Lanfredi, G.P.; et al. Host retromer protein sorting nexin 2 interacts with human respiratory syncytial virus structural proteins and is required for efficient viral production. MBio 2020, 11. [Google Scholar] [CrossRef]
- Vanover, D.; Smith, D.V.; Blanchard, E.L.; Alonas, E.; Kirschman, J.L.; Lifland, A.W.; Zurla, C.; Santangelo, P.J. RSV glycoprotein and genomic RNA dynamics reveal filament assembly prior to the plasma membrane. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- San-Juan-Vergara, H.; Sampayo-Escobar, V.; Reyes, N.; Cha, B.; Pacheco-Lugo, L.; Wong, T.; Peeples, M.E.; Collins, P.L.; Castaño, M.E.; Mohapatra, S.S. Cholesterol-Rich Microdomains as Docking Platforms for Respiratory Syncytial Virus in Normal Human Bronchial Epithelial Cells. J. Virol. 2012, 86, 1832–1843. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.Y.; Crowe, J.E. Molecular mechanisms driving respiratory syncytial virus assembly. Future Microbiol. 2013, 8, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.Y.; Cox, R.G.; Lifland, A.W.; Hotard, A.L.; Williams, J.V.; Moore, M.L.; Santangelo, P.J.; Crowe, J.E. A Critical Phenylalanine Residue in the Respiratory Syncytial Virus Fusion Protein Cytoplasmic Tail Mediates Assembly of Internal Viral Proteins into Viral Filaments and Particles. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Utley, T.J.; Ducharme, N.A.; Varthakavi, V.; Shepherd, B.E.; Santangelo, P.J.; Lindquist, M.E.; Goldenring, J.R.; Crowe, J.E. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc. Natl. Acad. Sci. USA 2008, 105, 10209–10214. [Google Scholar] [CrossRef] [Green Version]
- Zhivaki, D.; Lemoine, S.; Lim, A.; Morva, A.; Vidalain, P.-O.; Schandene, L.; Casartelli, N.; Rameix-Welti, M.-A.; Hervé, P.-L.; Dériaud, E.; et al. Respiratory Syncytial Virus Infects Regulatory B Cells in Human Neonates via Chemokine Receptor CX3CR1 and Promotes Lung Disease Severity. Immunity 2017, 46, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Amorim, N.A.; Da Silva, E.M.L.; De Castro, R.O.; Da Silva-Januário, M.E.; Mendonça, L.M.; Bonifacino, J.S.; Da Costa, L.J.; DaSilva, L.L.P. Interaction of HIV-1 nef protein with the host protein Alix promotes lysosomal targeting of cd4 receptor. J. Biol. Chem. 2014, 289, 27744–27756. [Google Scholar] [CrossRef] [Green Version]
- Covés-Datson, E.M.; King, S.R.; Legendre, M.; Gupta, A.; Chan, S.M.; Gitlin, E.; Kulkarni, V.V.; García, J.P.; Smee, D.F.; Lipka, E.; et al. A molecularly engineered antiviral banana lectin inhibits fusion and is efficacious against influenza virus infection in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 2122–2132. [Google Scholar] [CrossRef] [Green Version]
- Mruk, D.D.; Cheng, C.Y. Enhanced chemiluminescence (ECL) for routine immunoblotting. An inexpensive alternative to commercially available kits. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, P.J.; Bao, G. Dynamics of filamentous viral RNPs prior to egress. Nucleic Acids Res. 2007, 35, 3602–3611. [Google Scholar] [CrossRef] [Green Version]
- Banting, G.; Maile, R.; Roquemore, E.P. The steady state distribution of humTGN46 is not significantly altered in cells defective in clathrin-mediated endocytosis. J. Cell Sci. 1998, 111, 3451–3458. [Google Scholar]
- Chia, P.Z.C.; Gunn, P.; Gleeson, P.A. Cargo trafficking between endosomes and the trans-Golgi network. Histochem. Cell Biol. 2013, 140, 307–315. [Google Scholar] [CrossRef]
- Progida, C.; Bakke, O. Bidirectional traffic between the Golgi and the endosomes–machineries and regulation. J. Cell Sci. 2016, 129, 3971–3982. [Google Scholar] [CrossRef] [Green Version]
- Rojas, R.; Kametaka, S.; Haft, C.R.; Bonifacino, J.S. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell Biol. 2007, 27, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Haft, C.R.; Sierra, M.d.L.L.; Bafford, R.; Lesniak, M.A.; Barr, V.A.; Taylor, S.I. Human Orthologs of Yeast Vacuolar Protein Sorting Proteins Vps26, 29, and 35: Assembly into Multimeric Complexes. Mol. Biol Cell. 2000, 11, 4105–4116. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.; Rixon, H.W.M.; Sugrue, R.J. Respiratory syncytial virus assembly occurs in GM1-rich regions of the host-cell membrane and alters the cellular distribution of tyrosine phosphorylated caveolin-1. J. Gen. Virol. 2002, 83, 1841–1850. [Google Scholar] [CrossRef]
- Rivera-Toledo, E.; Gómez, B. Respiratory syncytial virus persistence in macrophages alters the profile of cellular gene expression. Viruses 2012, 4, 3270–3280. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.A.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M. Respiratory Syncytial Virus Uses CX3CR1 as a Receptor on Primary Human Airway Epithelial Cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef] [Green Version]
- Lingemann, M.; Mccarty, T.; Liu, X.; Buchholz, U.J.; Surman, S.; Martin, S.E.; Collins, P.L.; Munir, S. The alpha-1 subunit of the Na+,K+-ATPase (ATP1A1) is required for macropinocytic entry of respiratory syncytial virus (RSV) in human respiratory epithelial cells. PLoS Pathog. 2019, 15, e1007963. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.W.; Aberg, E.; Lawetz, C.; Delong, R.; Whitehead, P.; Liuzzi, M. Interaction between human respiratory syncytial virus (RSV) M2-1 and P proteins is required for reconstitution of M2-1-dependent RSV minigenome activity. J. Virol. 2003, 77, 10670–10676. [Google Scholar] [CrossRef] [Green Version]
- Bailly, B.; Richard, C.A.; Sharma, G.; Wang, L.; Johansen, L.; Cao, J.; Pendharkar, V.; Sharma, D.-C.; Galloux, M.; Wang, Y.; et al. Targeting human respiratory syncytial virus transcription anti-termination factor M2-1 to inhibit in vivo viral replication. Sci. Rep. 2016, 6, 25806. [Google Scholar] [CrossRef]
- Blondot, M.-L.; Dubosclard, V.; Fix, J.; Lassoued, S.; Aumont-Nicaise, M.; Bontems, F.; Eléouët, J.-F.; Sizun, C. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog. 2012, 8, e1002734. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza Cardoso, R.; Viana, R.M.M.; Vitti, B.C.; Coelho, A.C.L.; de Jesus, B.L.S.; de Paula Souza, J.; Pontelli, M.C.; Murakami, T.; Ventura, A.M.; Ono, A.; et al. Human Respiratory Syncytial Virus Infection in a Human T Cell Line Is Hampered at Multiple Steps. Viruses 2021, 13, 231. https://doi.org/10.3390/v13020231
de Souza Cardoso R, Viana RMM, Vitti BC, Coelho ACL, de Jesus BLS, de Paula Souza J, Pontelli MC, Murakami T, Ventura AM, Ono A, et al. Human Respiratory Syncytial Virus Infection in a Human T Cell Line Is Hampered at Multiple Steps. Viruses. 2021; 13(2):231. https://doi.org/10.3390/v13020231
Chicago/Turabian Stylede Souza Cardoso, Ricardo, Rosa Maria Mendes Viana, Brenda Cristina Vitti, Ana Carolina Lunardello Coelho, Bruna Laís Santos de Jesus, Juliano de Paula Souza, Marjorie Cornejo Pontelli, Tomoyuki Murakami, Armando Morais Ventura, Akira Ono, and et al. 2021. "Human Respiratory Syncytial Virus Infection in a Human T Cell Line Is Hampered at Multiple Steps" Viruses 13, no. 2: 231. https://doi.org/10.3390/v13020231
APA Stylede Souza Cardoso, R., Viana, R. M. M., Vitti, B. C., Coelho, A. C. L., de Jesus, B. L. S., de Paula Souza, J., Pontelli, M. C., Murakami, T., Ventura, A. M., Ono, A., & Arruda, E. (2021). Human Respiratory Syncytial Virus Infection in a Human T Cell Line Is Hampered at Multiple Steps. Viruses, 13(2), 231. https://doi.org/10.3390/v13020231