
Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
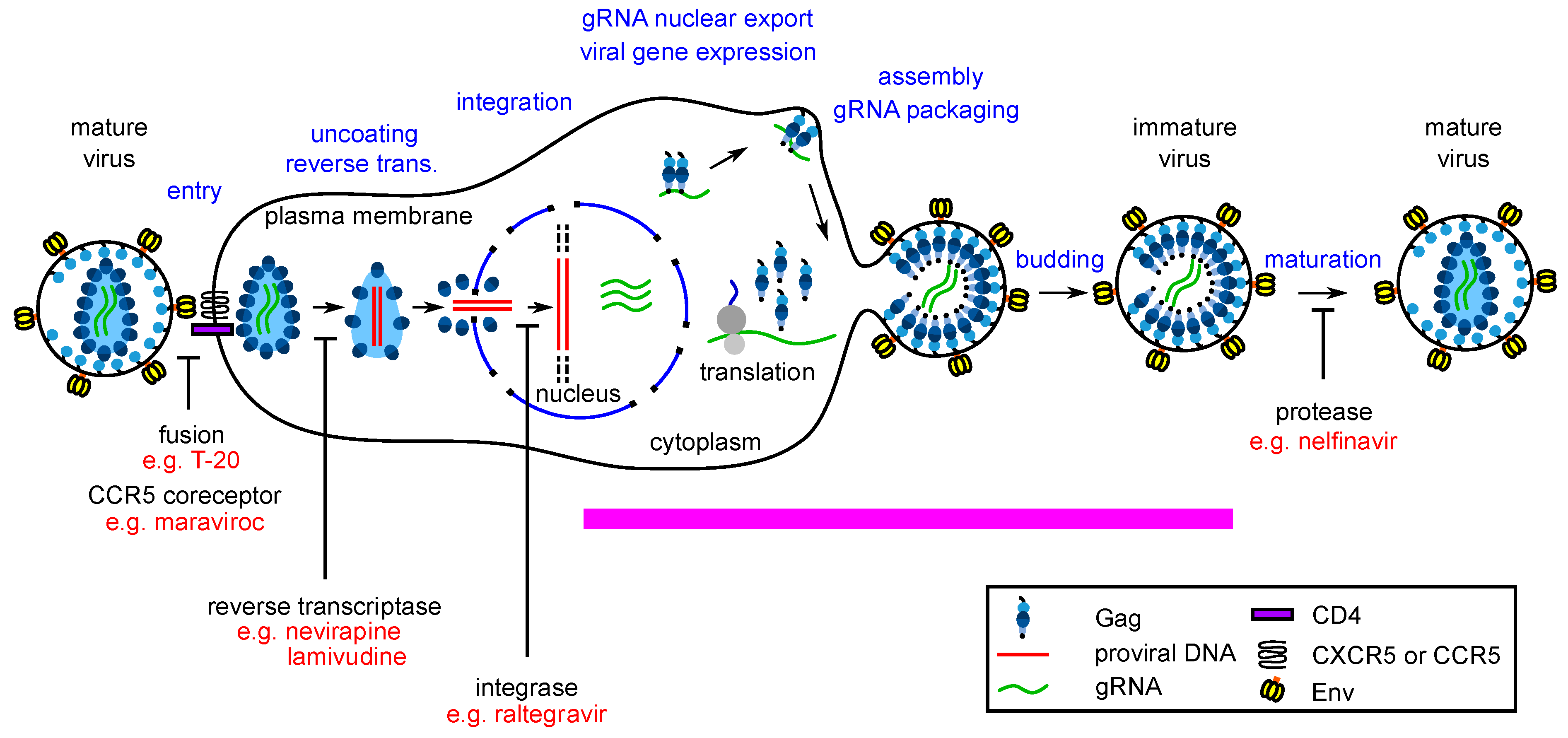
:1. Host-Targeting Antiviral Drugs—A Way to Fill a Gap in the Antiretroviral Armamentarium and Reduce the Specter of Antiviral Drug Resistance?
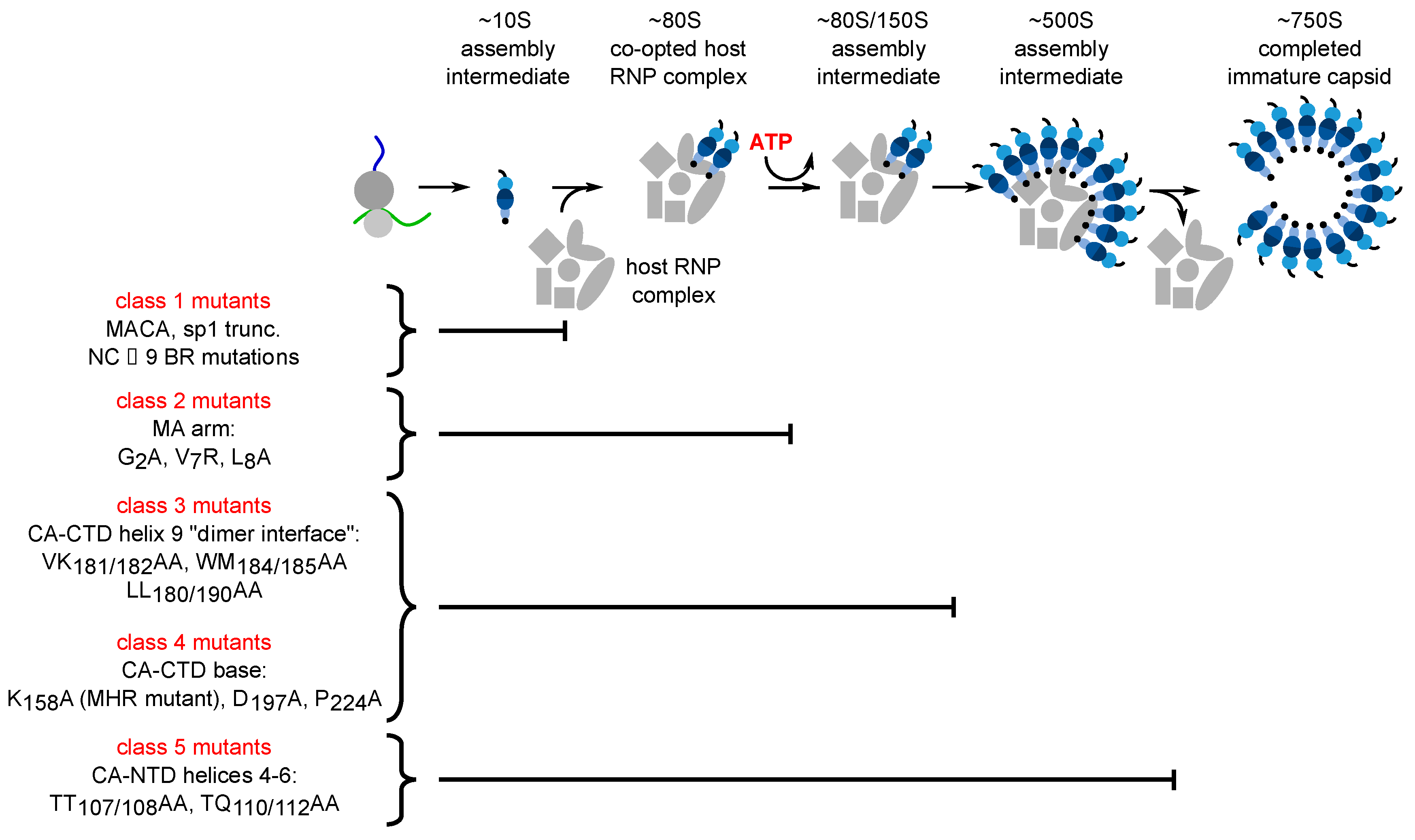
2. Spontaneous Assembly or Host-Catalyzed Assembly of HIV-1 Gag? Two Models with Implications for Assembly Inhibitors

3. Drugs Screens Over Two Decades Have Identified Small Molecule Inhibitors That Bind to CA
4. Screening for Host-Targeting Antiretroviral Compounds Using the Approach That Identified the First Rabies Virus Inhibitor
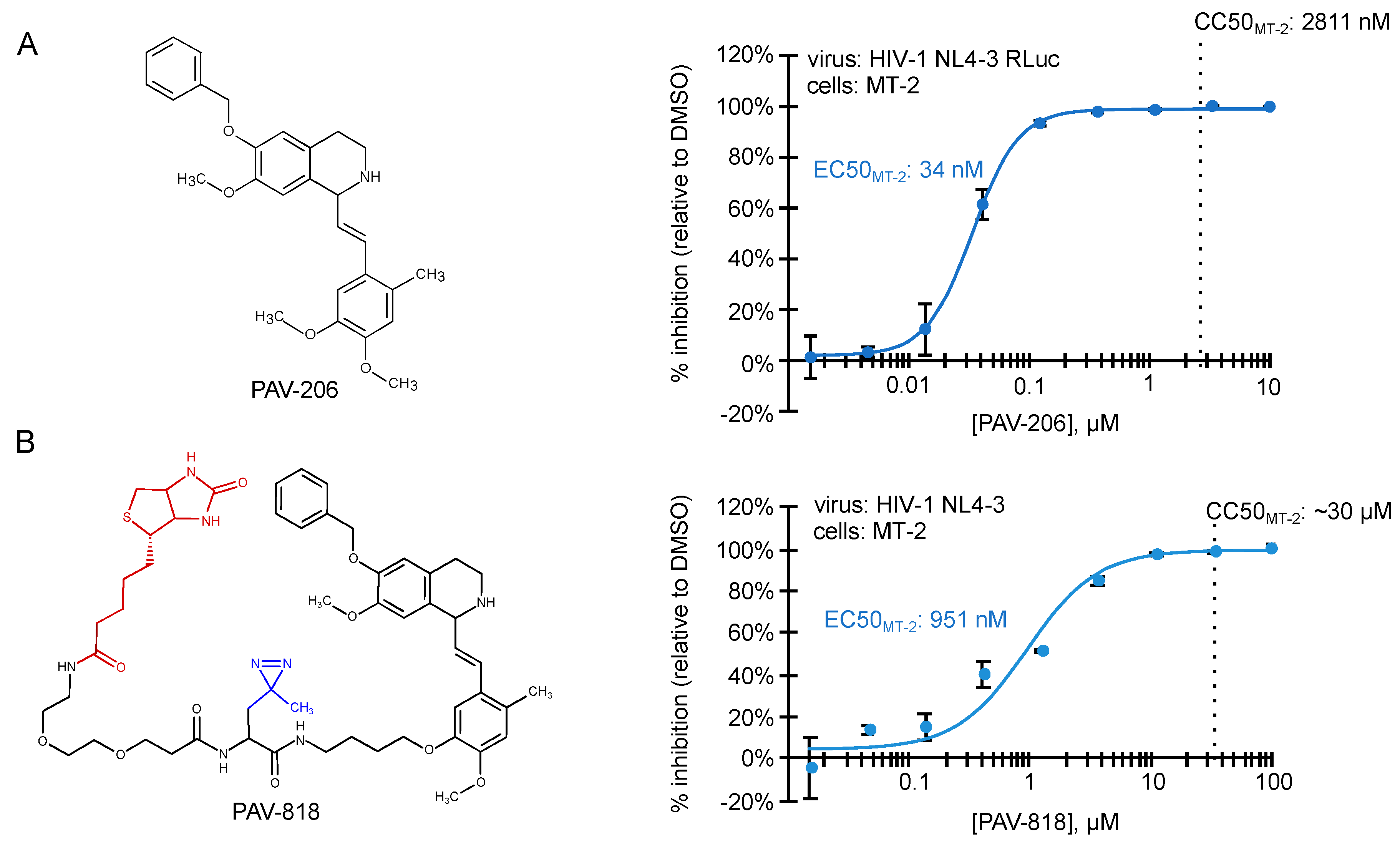
5. Identification of PAV-206, a Potent Small Molecule Inhibitor of HIV-1 Immature Capsid Assembly
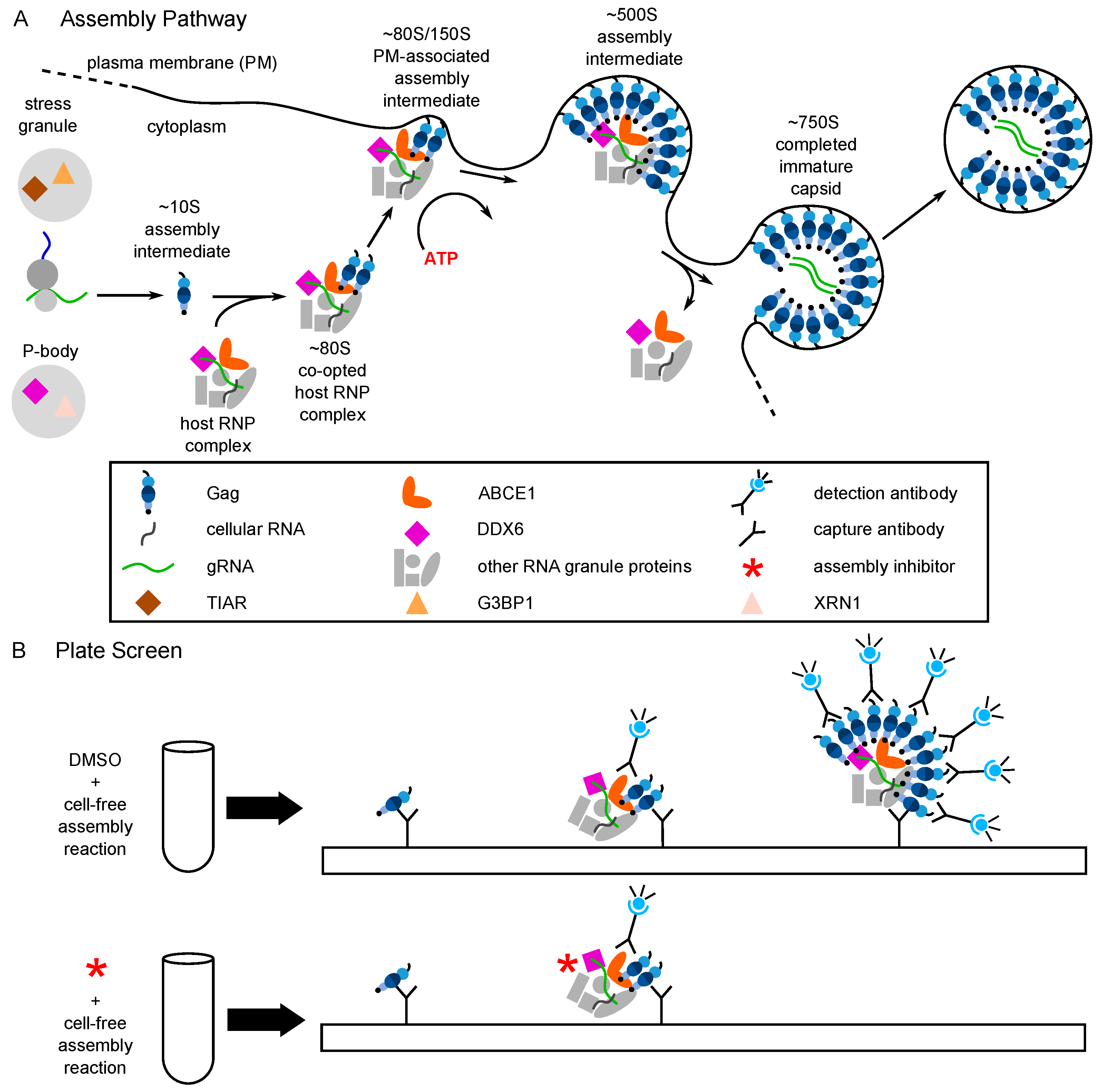
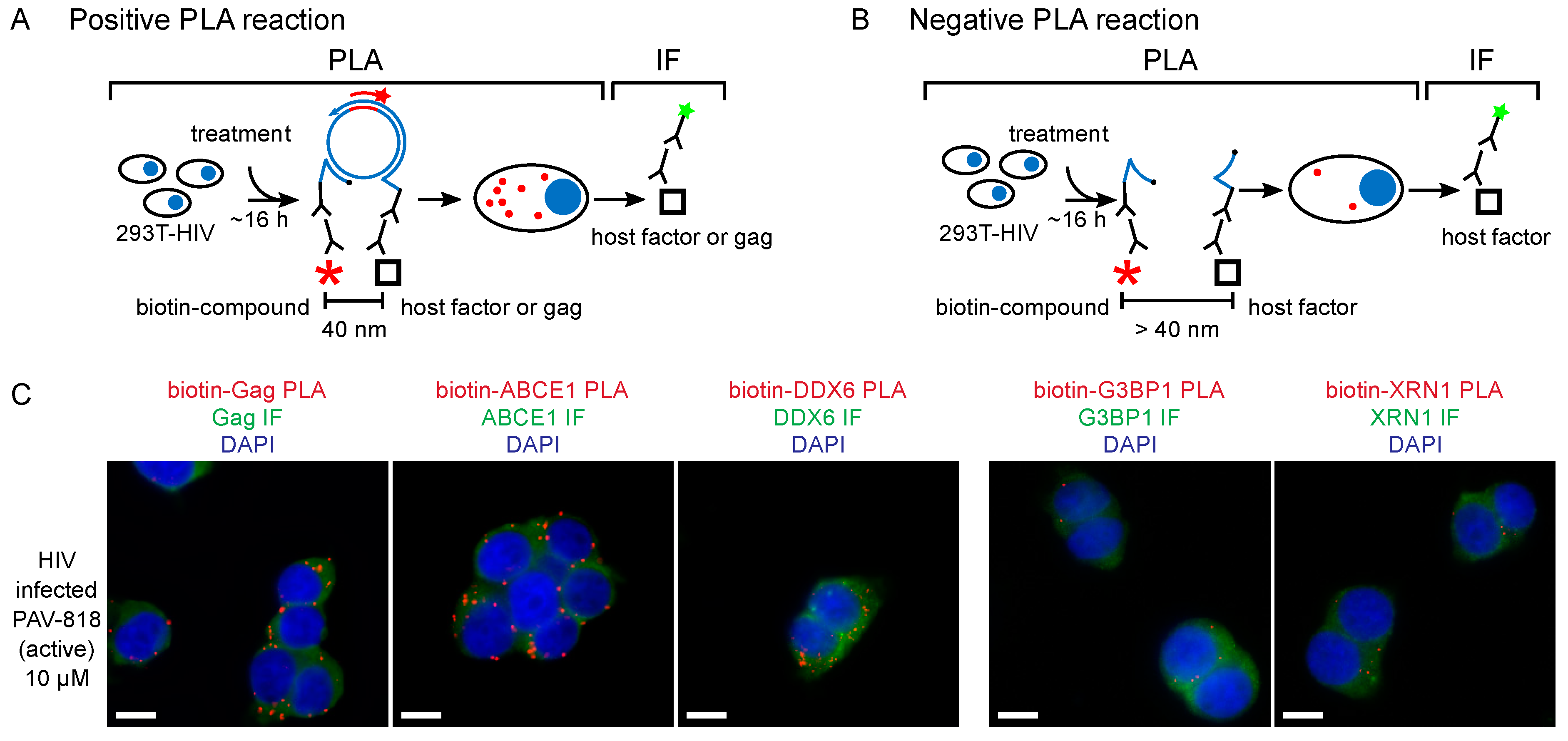
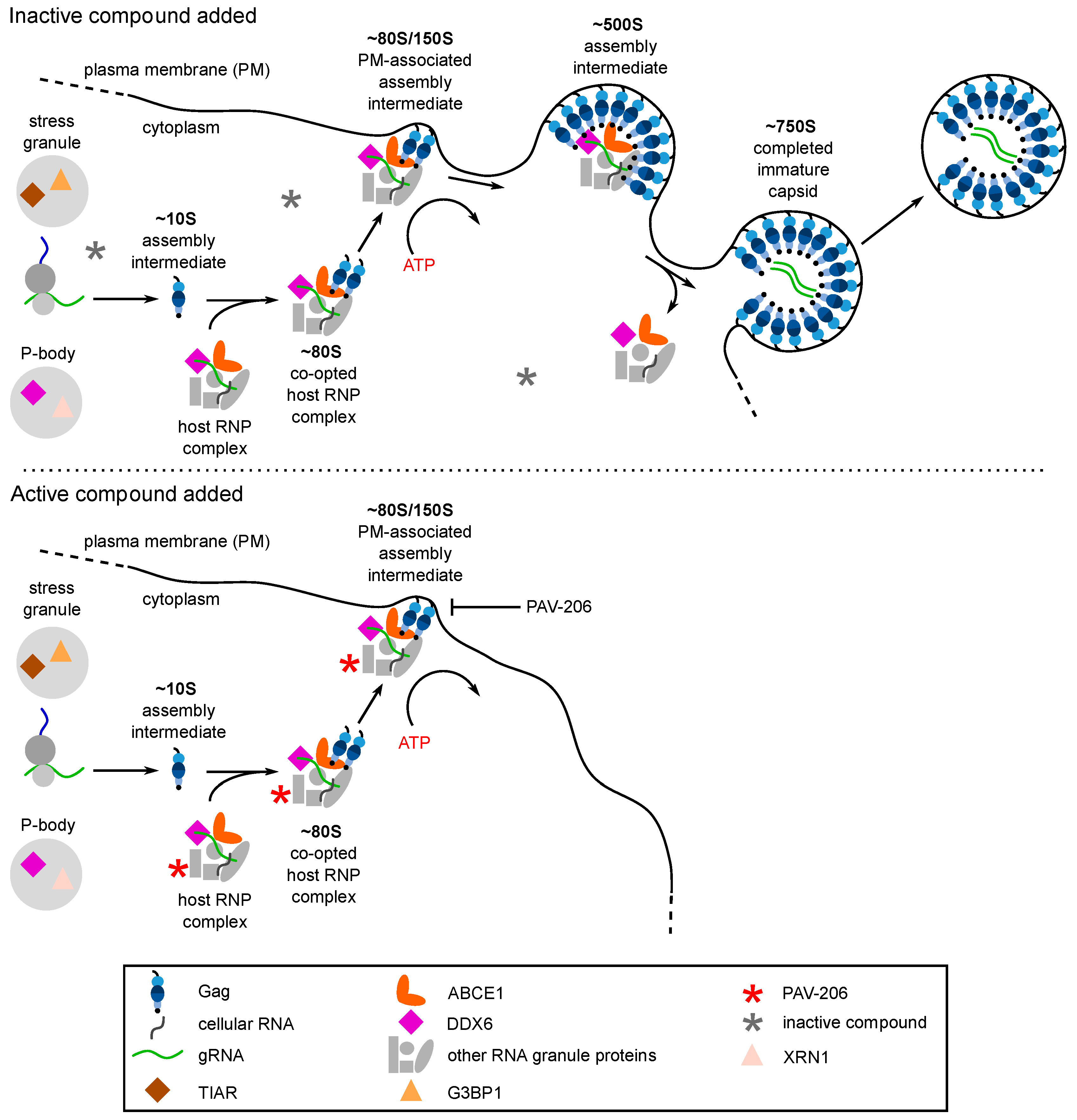
6. Evidence That PAV-206 Acts by Targeting Host Complexes Critical for HIV-1 Assembly
7. Host-Targeted Assembly Inhibitors—Challenges and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—2020 Fact Sheet. 2020. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 19 January 2021).
- Collier, D.A.; Monit, C.; Gupta, R.K. The Impact of HIV-1 Drug Escape on the Global Treatment Landscape. Cell Host Microbe 2019, 26, 48–60. [Google Scholar] [CrossRef]
- Boender, T.S.; Sigaloff, K.C.; McMahon, J.H.; Kiertiburanakul, S.; Jordan, M.R.; Barcarolo, J.; Ford, N.; Rinke de Wit, T.F.; Bertagnolio, S. Long-term Virological Outcomes of First-Line Antiretroviral Therapy for HIV-1 in Low- and Middle-Income Countries: A Systematic Review and Meta-analysis. Clin. Infect. Dis. 2015, 61, 1453–1461. [Google Scholar] [CrossRef]
- Gregson, J.; Kaleebu, P.; Marconi, V.C.; van Vuuren, C.; Ndembi, N.; Hamers, R.L.; Kanki, P.; Hoffmann, C.J.; Lockman, S.; Pillay, D.; et al. Occult HIV-1 drug resistance to thymidine analogues following failure of first-line tenofovir combined with a cytosine analogue and nevirapine or efavirenz in sub Saharan Africa: A retrospective multi-centre cohort study. Lancet Infect. Dis. 2017, 17, 296–304. [Google Scholar] [CrossRef] [Green Version]
- TenoRes Study, G. Global epidemiology of drug resistance after failure of WHO recommended first-line regimens for adult HIV-1 infection: A multicentre retrospective cohort study. Lancet Infect. Dis. 2016, 16, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: A systematic review and meta-regression analysis. Lancet Infect. Dis. 2018, 18, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Laborde-Balen, G.; Taverne, B.; Ndour, C.T.; Kouanfack, C.; Peeters, M.; Ndoye, I.; Delaporte, E. The fourth HIV epidemic. Lancet Infect. Dis. 2018, 18, 379–380. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Kassaye, S.G.; Barrow, G.; Sundaramurthi, J.C.; Jordan, M.R.; Shafer, R.W. HIV-1 transmitted drug resistance surveillance: Shifting trends in study design and prevalence estimates. J. Int. AIDS Soc. 2020, 23, e25611. [Google Scholar] [CrossRef]
- HIVinfo.NIH.gov. FDA-Approved HIV Medicine. 2020. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 19 January 2021).
- Preston, B.D.; Poiesz, B.J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase. Science 1988, 242, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.W.; Shafer, R.W. HIV-1 antiretroviral resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [Green Version]
- Miao, M.; De Clercq, E.; Li, G. Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Selvarajah, S.; Lingappa, A.F.; Michon, M.; Yu, S.F.; Macieik, A.; Mallesh, S.; Appaiah, U.; Crabtree, J.; Copeland, K.; Lin, J.; et al. From COVID-19 to the Common Cold: Novel Host-Targeted, Pan-Respiratory Antiviral Small Molecule Therapeutics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hendrix, C.W.; Collier, A.C.; Lederman, M.M.; Schols, D.; Pollard, R.B.; Brown, S.; Jackson, J.B.; Coombs, R.W.; Glesby, M.J.; Flexner, C.W.; et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2004, 37, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Hardy, W.D.; Gulick, R.M.; Mayer, H.; Fatkenheuer, G.; Nelson, M.; Heera, J.; Rajicic, N.; Goodrich, J. Two-year safety and virologic efficacy of maraviroc in treatment-experienced patients with CCR5-tropic HIV-1 infection: 96-week combined analysis of MOTIVATE 1 and 2. J. Acquir. Immune Defic. Syndr. 2010, 55, 558–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, R.M.; Fatkenheuer, G.; Burnside, R.; Hardy, W.D.; Nelson, M.R.; Goodrich, J.; Mukwaya, G.; Portsmouth, S.; Heera, J.R. Five-year safety evaluation of maraviroc in HIV-1-infected treatment-experienced patients. J. Acquir. Immune Defic. Syndr. 2014, 65, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Weehuizen, J.M.; Wensing, A.M.J.; Mudrikova, T.; Wit, F.; Hoepelman, A.I.M. Efficacy and safety of long-term maraviroc use in a heterogeneous group of HIV-infected patients: A retrospective cohort study. Int. J. Antimicrob. Agents 2019, 54, 215–222. [Google Scholar] [CrossRef]
- Francisci, D.; Pirro, M.; Schiaroli, E.; Mannarino, M.R.; Cipriani, S.; Bianconi, V.; Alunno, A.; Bagaglia, F.; Bistoni, O.; Falcinelli, E.; et al. Maraviroc Intensification Modulates Atherosclerotic Progression in HIV-Suppressed Patients at High Cardiovascular Risk. A Randomized, Crossover Pilot Study. Open Forum Infect. Dis. 2019, 6, ofz112. [Google Scholar] [CrossRef] [PubMed]
- Piconi, S.; Pocaterra, D.; Rainone, V.; Cossu, M.; Masetti, M.; Rizzardini, G.; Clerici, M.; Trabattoni, D. Maraviroc Reduces Arterial Stiffness in PI-Treated HIV-infected Patients. Sci. Rep. 2016, 6, 28853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnes, S.K.; Sheehan, J.H.; Aiken, C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutluay, S.B.; Bieniasz, P.D. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 2010, 6, e1001200. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr. Opin. Virol. 2019, 36, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Aiken, C. HIV-1 uncoating: Connection to nuclear entry and regulation by host proteins. Virology 2014, 454–455, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Solas, D.; Kitaygorodskyy, A.; Freeman, B.; Ressler, D.T.B.; Phuong, D.J.; Swain, J.V.; Matlack, K.; Hurt, C.R.; Lingappa, V.R.; et al. Identification of an Antiretroviral Small Molecule That Appears To Be a Host-Targeting Inhibitor of HIV-1 Assembly. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Bush, D.L.; Vogt, V.M. In Vitro Assembly of Retroviruses. Annu. Rev. Virol. 2014, 1, 561–580. [Google Scholar] [CrossRef]
- Dick, R.A.; Mallery, D.L.; Vogt, V.M.; James, L.C. IP6 Regulation of HIV Capsid Assembly, Stability, and Uncoating. Viruses 2018, 10, 640. [Google Scholar] [CrossRef] [Green Version]
- Rein, A.; Datta, S.A.; Jones, C.P.; Musier-Forsyth, K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem. Sci. 2011, 36, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Bieniasz, P.; Telesnitsky, A. Multiple, Switchable Protein:RNA Interactions Regulate Human Immunodeficiency Virus Type 1 Assembly. Annu. Rev. Virol. 2018, 5, 165–183. [Google Scholar] [CrossRef]
- Rein, A. RNA Packaging in HIV. Trends Microbiol. 2019, 27, 715–723. [Google Scholar] [CrossRef]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA 2001, 98, 10875–10879. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.; Rein, A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 1999, 73, 2270–2279. [Google Scholar] [CrossRef] [Green Version]
- Lingappa, J.R.; Hill, R.L.; Wong, M.L.; Hegde, R.S. A multistep, ATP-dependent pathway for assembly of human immunodeficiency virus capsids in a cell-free system. J. Cell Biol. 1997, 136, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, J.R.; Newman, M.A.; Klein, K.C.; Dooher, J.E. Comparing capsid assembly of primate lentiviruses and hepatitis B virus using cell-free systems. Virology 2005, 333, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooher, J.E.; Schneider, B.L.; Reed, J.C.; Lingappa, J.R. Host ABCE1 is at Plasma Membrane HIV Assembly Sites and Its Dissociation from Gag is Linked to Subsequent Events of Virus Production. Traffic 2007, 8, 195–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingappa, J.R.; Dooher, J.E.; Newman, M.A.; Kiser, P.K.; Klein, K.C. Basic residues in the nucleocapsid domain of Gag are required for interaction of HIV-1 gag with ABCE1 (HP68), a cellular protein important for HIV-1 capsid assembly. J. Biol. Chem. 2006, 281, 3773–3784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, B.A.; Reed, J.C.; Geary, C.D.; Swain, J.V.; Lingappa, J.R. A temporospatial map that defines specific steps at which critical surfaces in the Gag MA and CA domains act during immature HIV-1 capsid assembly in cells. J. Virol. 2014, 88, 5718–5741. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.R.; Hill, R.L.; Lingappa, J.R. Effect of mutations in Gag on assembly of immature human immunodeficiency virus type 1 capsids in a cell-free system. Virology 2001, 279, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Q.; Dong, J.; Ishimura, A.; Daar, I.; Hinnebusch, A.G.; Dean, M. The Essential Vertebrate ABCE1 Protein Interacts with Eukaryotic Initiation Factors. J. Biol. Chem. 2006, 281, 7452–7457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Quiles, C.; Mateo-Bonmati, E.; Micol, J.L. ABCE Proteins: From Molecules to Development. Front. Plant. Sci. 2018, 9, 1125. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, C.; Klein, K.C.; Kiser, P.K.; Singh, A.R.S.; Firestein, B.L.; Riba, S.C.; Lingappa, J.R. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature 2002, 415, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Molter, B.; Geary, C.D.; McNevin, J.; McElrath, J.; Giri, S.; Klein, K.C.; Lingappa, J.R. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J. Cell Biol. 2012, 198, 439–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingappa, J.R.; Reed, J.C.; Tanaka, M.; Chutiraka, K.; Robinson, B.A. How HIV-1 Gag assembles in cells: Putting together pieces of the puzzle. Virus Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Jarmoskaite, I.; Russell, R. RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 2014, 83, 697–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajas, B.C.; Tanaka, M.; Robinson, B.A.; Phuong, D.J.; Chutiraka, K.; Reed, J.C.; Lingappa, J.R. Identifying the assembly intermediate in which Gag first associates with unspliced HIV-1 RNA suggests a novel model for HIV-1 RNA packaging. PLoS Pathog. 2018, 14, e1006977. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Robinson, B.A.; Chutiraka, K.; Geary, C.D.; Reed, J.C.; Lingappa, J.R. Mutations of Conserved Residues in the Major Homology Region Arrest Assembling HIV-1 Gag as a Membrane-Targeted Intermediate Containing Genomic RNA and Cellular Proteins. J. Virol. 2015, 90, 1944–1963. [Google Scholar] [CrossRef] [Green Version]
- Jardine, P.J. Slow and steady wins the race: Physical limits on the rate of viral DNA packaging. Curr. Opin. Virol. 2019, 36, 32–37. [Google Scholar] [CrossRef]
- Deng, Y.; Hammond, J.A.; Pauszek, R.; Ozog, S.; Chai, I.; Rabuck-Gibbons, J.; Lamichhane, R.; Henderson, S.C.; Millar, D.P.; Torbett, B.E.; et al. Discrimination between Functional and Non-functional Cellular Gag Complexes involved in HIV-1 Assembly. J. Mol. Biol. 2021, 433, 166842. [Google Scholar] [CrossRef]
- Dooher, J.E.; Lingappa, J.R. Conservation of a step-wise, energy-sensitive pathway involving HP68 for assembly of primate lentiviral capsids in cells. J. Virol. 2004, 78, 1645–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C.; Westergreen, N.; Barajas, B.C.; Ressler, D.T.B.; Phuong, D.J.; Swain, J.V.; Lingappa, V.R.; Lingappa, J.R. The formation of RNA granule-derived capsid assembly intermediates appears to be conserved between HIV-1 and the non-primate lentivirus FIV. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, A.; Waheed, A.A.; Joshi, A.; Freed, E.O. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: Use of a novel Gag multimerization assay. J. Virol. 2005, 79, 14131–14140. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.A.; Simon, M.N.; Gross, I.; Krausslich, H.G.; Fuller, S.D.; Vogt, V.M.; Johnson, M.C. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 2004, 11, 672–675. [Google Scholar] [CrossRef]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Chazal, N.; Carriere, C.; Gay, B.; Boulanger, P. Phenotypic characterization of insertion mutants of the human immunodeficiency virus type 1 Gag precursor expressed in recombinant baculovirus-infected cells. J. Virol. 1994, 68, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheysen, D.; Jacobs, E.; de Foresta, F.; Thiriart, C.; Francotte, M.; Thines, D.; De Wilde, M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 1989, 59, 103–112. [Google Scholar] [CrossRef]
- O’Carroll, I.P.; Crist, R.M.; Mirro, J.; Harvin, D.; Soheilian, F.; Kamata, A.; Nagashima, K.; Rein, A. Functional redundancy in HIV-1 viral particle assembly. J. Virol. 2012, 86, 12991–12996. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Isaacson, J.; Patick, A.K.; Blair, W.S. High-throughput human immunodeficiency virus type 1 (HIV-1) full replication assay that includes HIV-1 Vif as an antiviral target. Antimicrob. Agents Chemother. 2005, 49, 3833–3841. [Google Scholar] [CrossRef] [Green Version]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; Buffone, C.; Opp, S.; Valle-Casuso, J.; Diaz-Griffero, F. BI-2 destabilizes HIV-1 cores during infection and Prevents Binding of CPSF6 to the HIV-1 Capsid. Retrovirology 2014, 11, 120. [Google Scholar] [CrossRef]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; Pornillos, O. Assembly and architecture of HIV. Adv. Exp. Med. Biol. 2012, 726, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Loeliger, E.; Kinde, I.; Kyere, S.; Mayo, K.; Barklis, E.; Sun, Y.; Huang, M.; Summers, M.F. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 2003, 327, 1013–1020. [Google Scholar] [CrossRef]
- Kelly, B.N.; Kyere, S.; Kinde, I.; Tang, C.; Howard, B.R.; Robinson, H.; Sundquist, W.I.; Summers, M.F.; Hill, C.P. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 2007, 373, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, C.T.; Titolo, S.; von Schwedler, U.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Faucher, A.M.; Coulombe, R.; Banik, S.S.; Fader, L.; et al. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 2012, 86, 6643–6655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thenin-Houssier, S.; de Vera, I.M.; Pedro-Rosa, L.; Brady, A.; Richard, A.; Konnick, B.; Opp, S.; Buffone, C.; Fuhrmann, J.; Kota, S.; et al. Ebselen, a Small-Molecule Capsid Inhibitor of HIV-1 Replication. Antimicrob. Agents Chemother. 2016, 60, 2195–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sonnerborg, A. GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds. Front. Microbiol. 2019, 10, 1227. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384. [Google Scholar] [CrossRef]
- Lingappa, U.F.; Wu, X.; Macieik, A.; Yu, S.F.; Atuegbu, A.; Corpuz, M.; Francis, J.; Nichols, C.; Calayag, A.; Shi, H.; et al. Host-rabies virus protein-protein interactions as druggable antiviral targets. Proc. Natl. Acad. Sci. USA 2013, 110, E861–E868. [Google Scholar] [CrossRef] [Green Version]
- Soderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Marcelin, A.G.; Charpentier, C.; Jary, A.; Perrier, M.; Margot, N.; Callebaut, C.; Calvez, V.; Descamps, D. Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients. J. Antimicrob. Chemother. 2020, 75, 1588–1590. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Perry, C.M.; Frampton, J.E.; McCormack, P.L.; Siddiqui, M.A.; Cvetkovic, R.S. Nelfinavir: A review of its use in the management of HIV infection. Drugs 2005, 65, 2209–2244. [Google Scholar] [CrossRef]
- Garbelli, A.; Riva, V.; Crespan, E.; Maga, G. How to win the HIV-1 drug resistance hurdle race: Running faster or jumping higher? Biochem. J. 2017, 474, 1559–1577. [Google Scholar] [CrossRef]
- Eakle, R.; Venter, F.; Rees, H. Pre-exposure prophylaxis (PrEP) in an era of stalled HIV prevention: Can it change the game? Retrovirology 2018, 15, 29. [Google Scholar] [CrossRef] [Green Version]
- Nugent, D.; Gilson, R. Where next with preexposure prophylaxis? Curr. Opin. Infect. Dis. 2017, 30, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Multitalented actors inside and outside the cell: Recent discoveries add to the number of moonlighting proteins. Biochem. Soc. Trans. 2019, 47, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, R.; Muller-Schiffmann, A.; Bader, V.; Selvarajah, S.; Dey, D.; Lingappa, V.R.; Korth, C. Viral capsid assembly as a model for protein aggregation diseases: Active processes catalyzed by cellular assembly machines comprising novel drug targets. Virus Res. 2015, 207, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Muller-Schiffmann, A.; Trossbach, S.V.; Lingappa, V.R.; Korth, C. Viruses as ’Truffle Hounds’: Molecular Tools for Untangling Brain Cellular Pathology. Trends Neurosci. 2020. [Google Scholar] [CrossRef]
- Campillos, M.; Doerks, T.; Shah, P.K.; Bork, P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006, 22, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Campioni, M.R.; Finkbeiner, S. Going retro: Ancient viral origins of cognition. Neuron 2015, 86, 346–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wu, J.; Ward, M.D.; Yang, S.; Chuang, Y.A.; Xiao, M.; Li, R.; Leahy, D.J.; Worley, P.F. Structural basis of arc binding to synaptic proteins: Implications for cognitive disease. Neuron 2015, 86, 490–500. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lingappa, J.R.; Lingappa, V.R.; Reed, J.C. Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses 2021, 13, 451. https://doi.org/10.3390/v13030451
Lingappa JR, Lingappa VR, Reed JC. Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses. 2021; 13(3):451. https://doi.org/10.3390/v13030451
Chicago/Turabian StyleLingappa, Jaisri R., Vishwanath R. Lingappa, and Jonathan C. Reed. 2021. "Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor" Viruses 13, no. 3: 451. https://doi.org/10.3390/v13030451
APA StyleLingappa, J. R., Lingappa, V. R., & Reed, J. C. (2021). Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses, 13(3), 451. https://doi.org/10.3390/v13030451