
Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics



Abstract
:1. Introduction
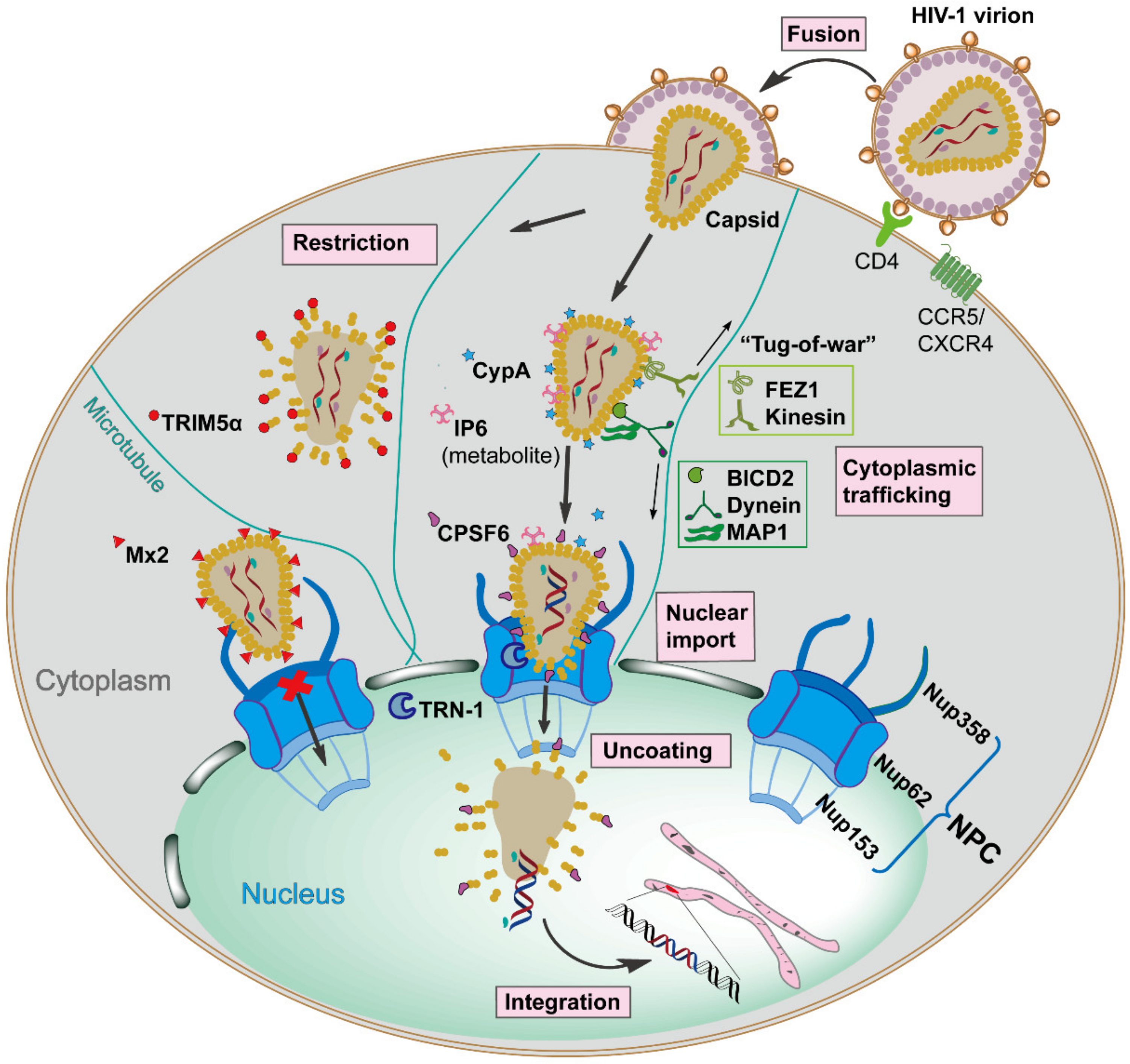
2. The Dynamic Capsid–Host Interactions during Early HIV-1 Infection
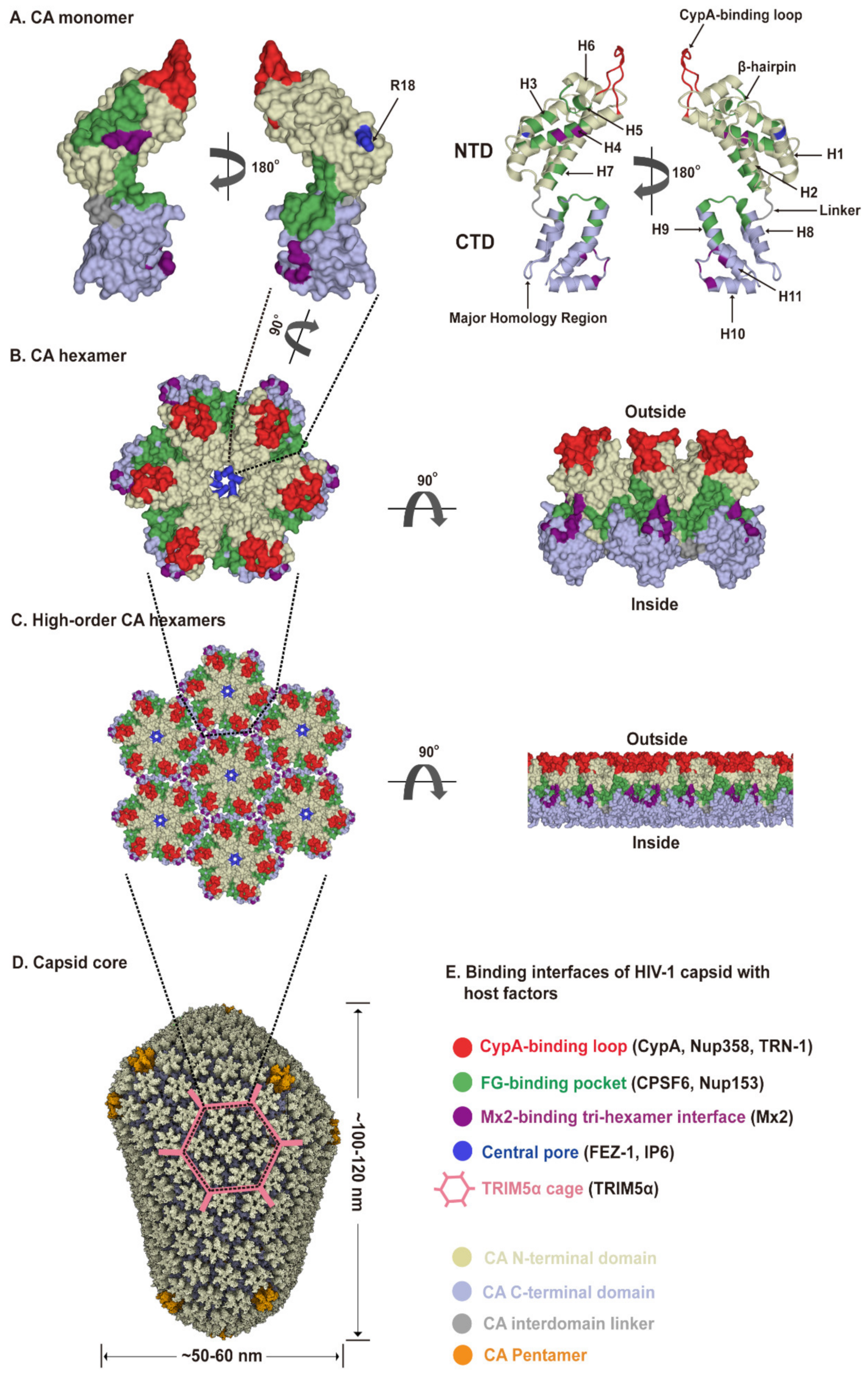
2.1. HIV-1 Capsid Architecture
2.2. Capsid-Host Factor Interactions Contribute to Trafficking and Capsid Stability in the Cytoplasm
2.3. Roles of Capsid in Nuclear Import
2.4. Capsid-Targeting Host Restriction Factors
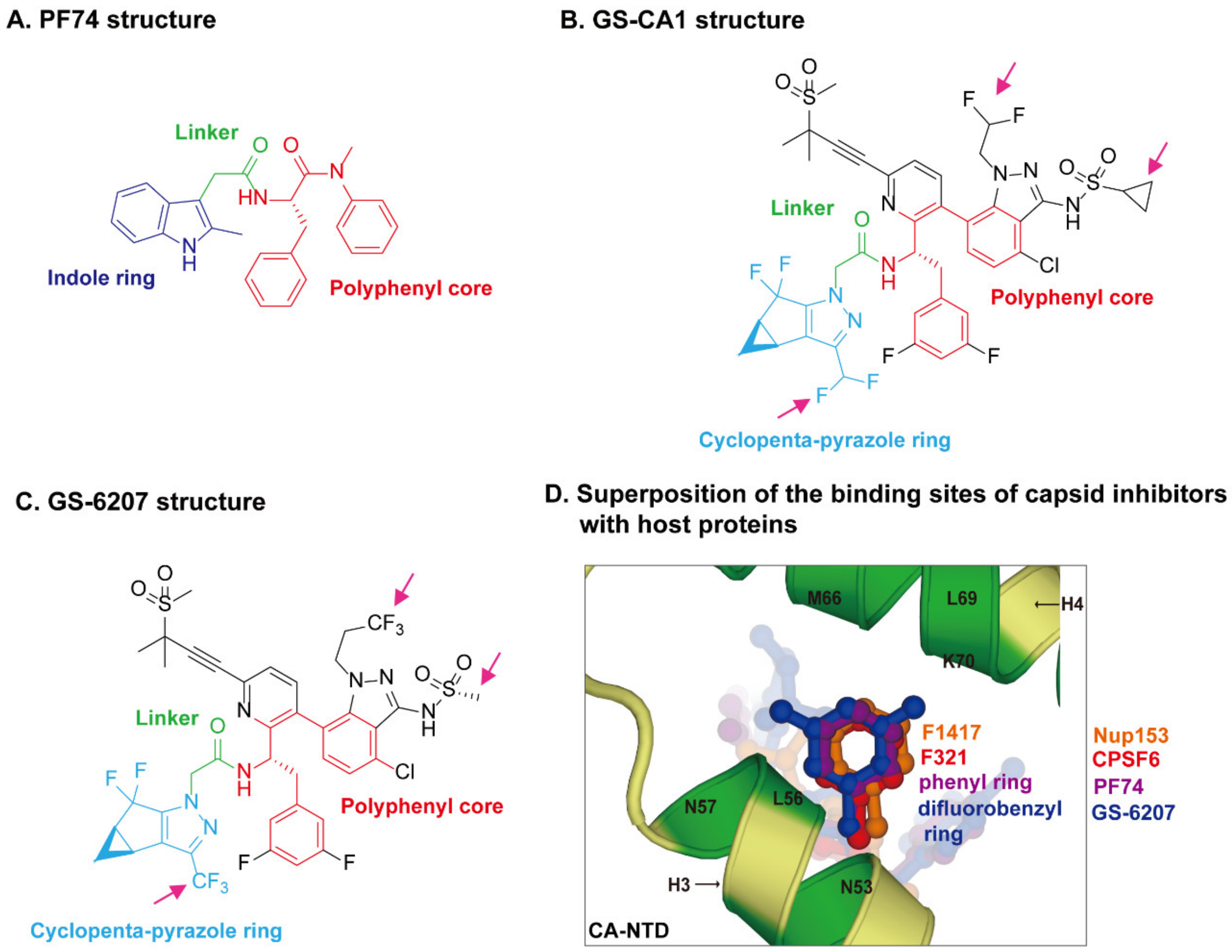
3. Recent Advances in HIV-1 Capsid Inhibitors
4. High-Throughput Screening Techniques Used to Reveal HIV-1–Host Interactions
4.1. Genomic Approaches
4.2. Proteomic Approaches
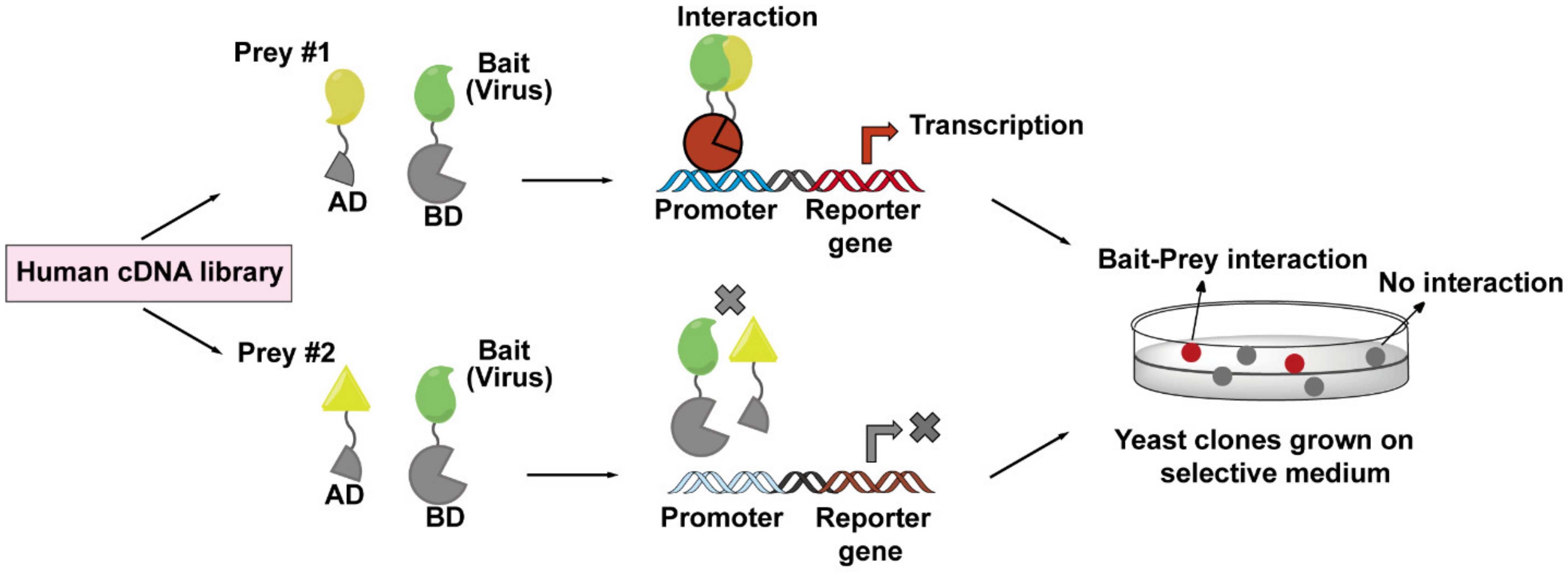
4.3. Yeast Two-Hybrid (Y2H) Screen
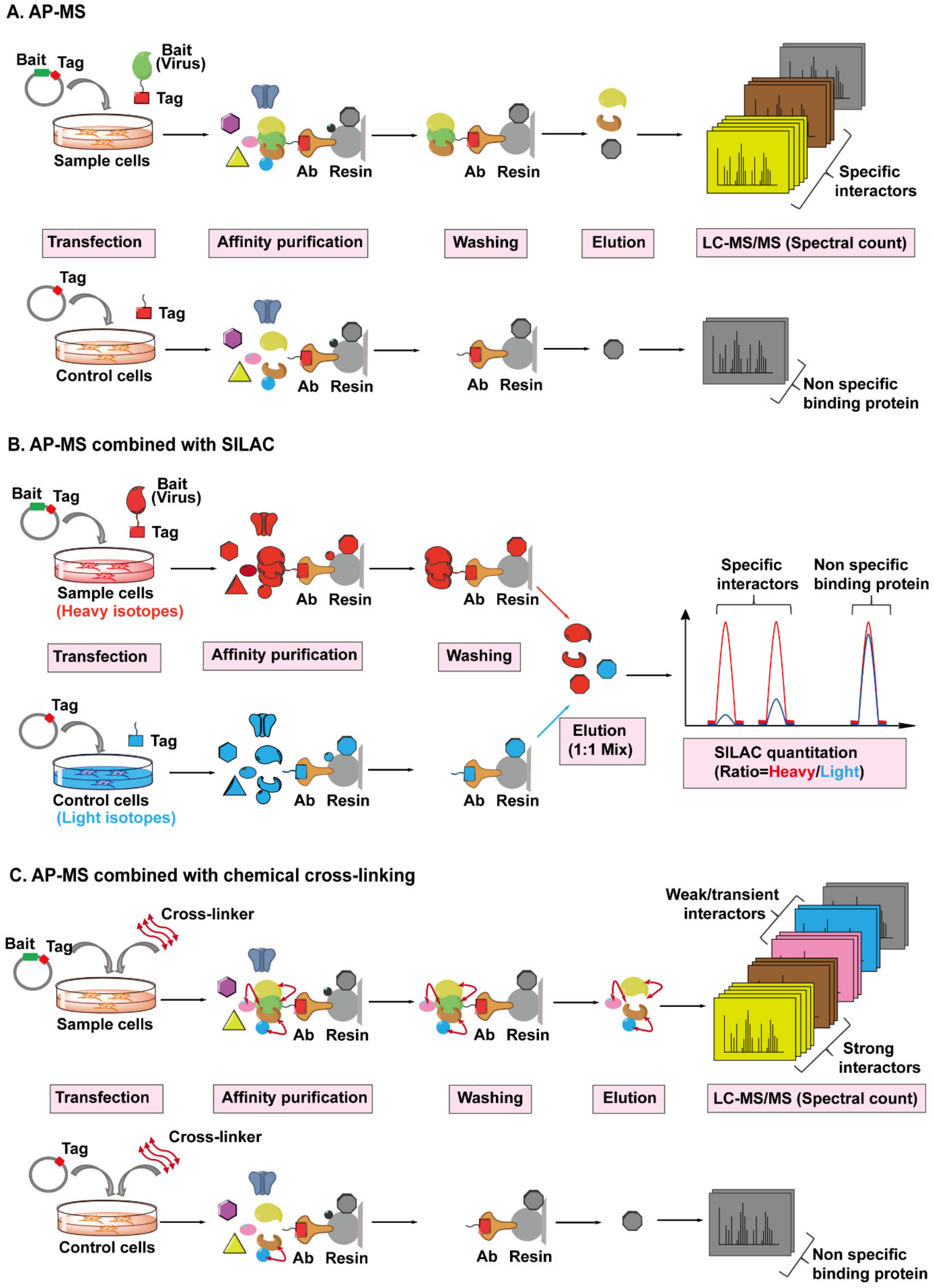
4.4. AP-MS Combined with Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) or Chemical Cross-Linking
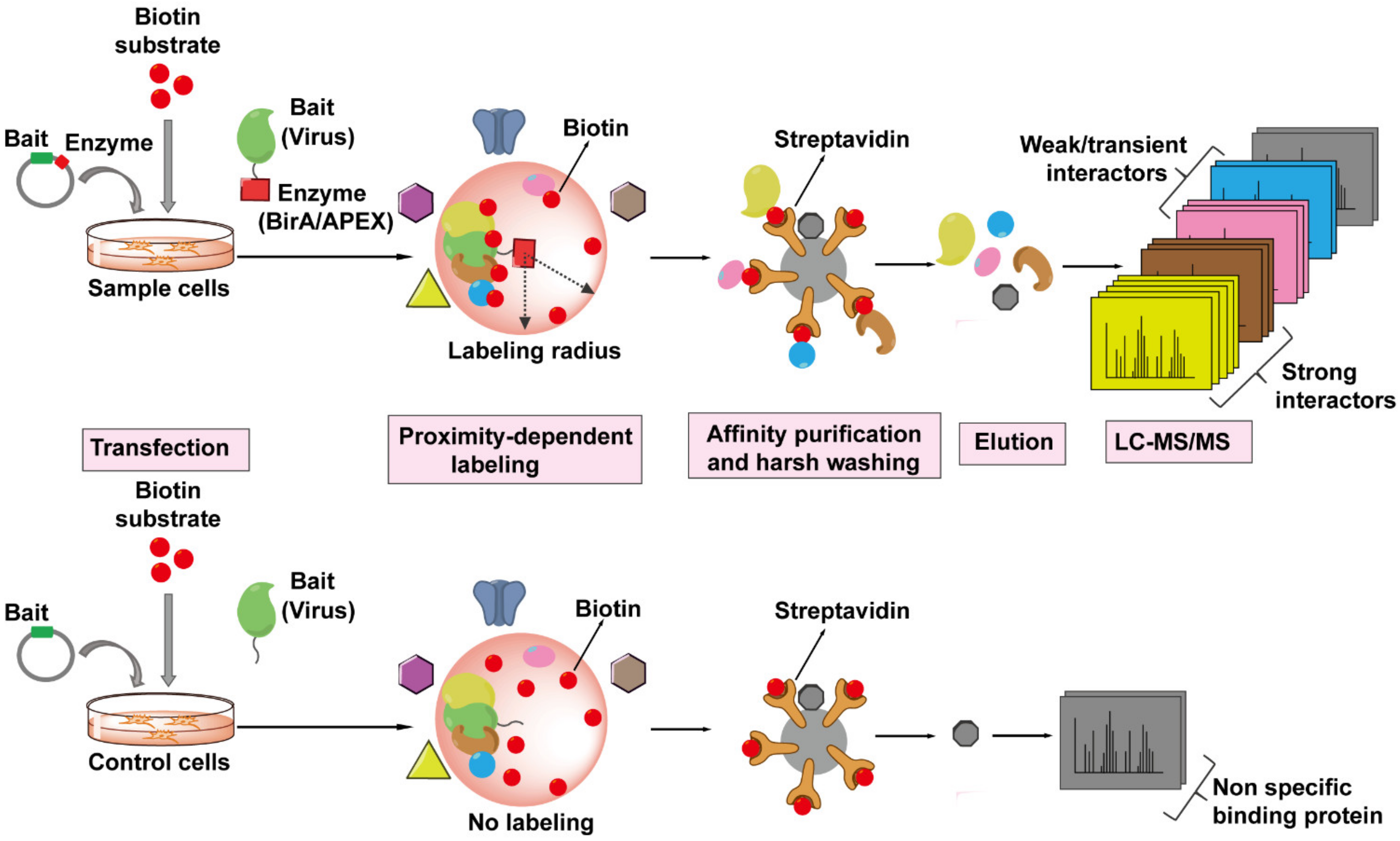
4.5. Proximity-Dependent Labeling (PDL) Technology
5. Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | acquired immune deficiency syndrome |
| AP-MS | affinity purification followed by mass spectrometry |
| ART | anti-retroviral therapy |
| BICD2 | Bicaudal D2 |
| CA | monomeric capsid protein |
| CFIm | cleavage factor I mammalian |
| CHD | CypA homologous domain |
| CPSF6 | Cleavage and polyadenylation specificity factor subunit 6 |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 |
| CypA | cyclophilin A |
| cyro-ET | cyro-electron tomography |
| DmrB | drug-inducible dimerization domain B |
| dNTPs | deoxynucleotide triphosphates |
| FDA | Food and Drug Administration |
| FEZ1 | Fasciculation and Elongation Protein Zeta 1 |
| HIV-1 | The Human Immunodeficiency Virus type 1 |
| IFNs | interferons |
| IN | integrase |
| IP6 | inositol hexaphosphate |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| MHR | major homology region |
| MT | microtubule |
| MX2 | myxovirus resistance 2 |
| NPC | nuclear pore complex |
| Nup | nucleoporin |
| PDL | proximity-dependent labeling |
| PIC | pre-integration complex |
| PPIase | peptidyl-prolyl cis-trans isomerase |
| PPIs | protein–protein interactions |
| RING | Really Interesting New Gene |
| RNAi | RNA interference |
| RT | reverse transcriptase |
| SILAC | Stable Isotope Labeling with Amino acids in Cell culture |
| siRNA | small-interfering RNA |
| SPRY domain | SPla and the RYanodine Receptor |
| TAP | tandem affinity purification |
| TRIM5α | tripartite motif-containing protein 5 alpha |
| TRN-1 | Transportin-1 |
| Y2H | yeast two-hybrid |
References
- Freed, E.O. HIV-1 replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860. [Google Scholar] [CrossRef]
- Swanstrom, R.; Coffin, J. HIV-1 pathogenesis: The virus. Cold Spring Harb. Perspect. Med. 2012, 2, a007443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, P.; Sahu, A.K.; Mishra, T.; Bhardwaj, V.; Chande, A. From entry to egress: Strategic exploitation of the cellular processes by HIV-1. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Chen, B. Molecular mechanism of HIV-1 entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Hehl, E.A.; Joshi, P.; Kalpana, G.V.; Prasad, V.R. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J. Virol. 2004, 78, 5056–5067. [Google Scholar] [CrossRef] [Green Version]
- Gaudin, R.; de Alencar, B.C.; Arhel, N.; Benaroch, P. HIV trafficking in host cells: Motors wanted! Trends Cell Biol. 2013, 23, 652–662. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turonova, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Engelman, A.N. Capsid-Dependent host factors in HIV-1 infection. Trends Microbiol. 2017, 25, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple roles of HIV-1 capsid during the virus replication cycle. Virol. Sin. 2019, 34, 119–134. [Google Scholar] [CrossRef] [Green Version]
- Temple, J.; Tripler, T.N.; Shen, Q.; Xiong, Y. A snapshot of HIV-1 capsid-host interactions. Curr. Res. Struct. Biol. 2020, 2. [Google Scholar] [CrossRef]
- Summers, B.J.; Digianantonio, K.M.; Smaga, S.S.; Huang, P.-T.; Zhou, K.; Gerber, E.E.; Wang, W.; Xiong, Y. Modular HIV-1 capsid assemblies reveal diverse host-capsid recognition mechanisms. Cell Host Microbe 2019, 26, 203–216.e6. [Google Scholar] [CrossRef]
- Broder, S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir. Res. 2010, 85, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Lu, T. High Active Anti-retroviral Therapy for HIV/AIDS, Progresses and Drawback. Adv. Pharmacoepidemiol. Drug Saf. 2012, 1, e115. [Google Scholar] [CrossRef] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Trovato, M.; D’Apice, L.; Prisco, A.; De Berardinis, P. HIV vaccination: A roadmap among advancements and concerns. Int. J. Mol. Sci. 2018, 19, 1241. [Google Scholar] [CrossRef] [Green Version]
- Fätkenheuer, G.; Pozniak, A.L.; Johnson, M.A.; Plettenberg, A.; Staszewski, S.; Hoepelman, A.I.; Saag, M.S.; Goebel, F.D.; Rockstroh, J.K.; Dezube, B.J. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 2005, 11, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnes, S.K.; Sheehan, J.H.; Aiken, C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359. [Google Scholar] [CrossRef]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef] [Green Version]
- McArthur, C.; Gallazzi, F.; Quinn, T.P.; Singh, K. HIV Capsid Inhibitors Beyond PF74. Diseases 2019, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364. [Google Scholar] [PubMed]
- Jaeger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365–370. [Google Scholar] [CrossRef]
- Ivanov, S.; Lagunin, A.; Filimonov, D.; Tarasova, O. Network-Based Analysis of OMICs Data to Understand the HIV-Host Interaction. Front. Microbiol. 2020. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- Mattei, S.; Glass, B.; Hagen, W.J.; Kräusslich, H.-G.; Briggs, J.A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef]
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 maturation: Lessons learned from inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D. Intracellular transport using microtubule-based motors. Annu. Rev. Cell Biol. 1987, 3, 347–378. [Google Scholar] [CrossRef]
- Dharan, A.; Opp, S.; Abdel-Rahim, O.; Keceli, S.K.; Imam, S.; Diaz-Griffero, F.; Campbell, E.M. Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. USA 2017, 114, E10707–E10716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnes, S.K.; Zhou, J.; Aiken, C. HIV-1 engages a dynein-dynactin-BICD2 complex for infection and transport to the nucleus. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Malikov, V.; Da Silva, E.S.; Jovasevic, V.; Bennett, G.; de Souza Aranha Vieira, D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Huang, P.-T.; Summers, B.J.; Xu, C.; Perilla, J.R.; Malikov, V.; Naghavi, M.H.; Xiong, Y. FEZ1 is recruited to a conserved cofactor site on capsid to promote HIV-1 trafficking. Cell Rep. 2019, 28, 2373–2385.e7. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Portilho, D.M.; Danckaert, A.; Munier, S.; Becker, A.; Roux, P.; Zambo, A.; Shorte, S.; Jacob, Y.; Vidalain, P.-O. Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J. Biol. Chem. 2015, 290, 4631–4646. [Google Scholar] [CrossRef] [Green Version]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Ni, T.; Gerard, S.; Zhao, G.; Dent, K.; Ning, J.; Zhou, J.; Shi, J.; Anderson-Daniels, J.; Li, W.; Jang, S. Intrinsic curvature of the HIV-1 CA hexamer underlies capsid topology and interaction with cyclophilin A. Nat. Struct. Mol. Biol. 2020, 27, 855–862. [Google Scholar] [CrossRef]
- Li, Y.; Kar, A.K.; Sodroski, J. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 2009, 83, 10951–10962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.B.; Shi, J.; Hout, D.R.; Oztop, I.; Krishnan, L.; Ahn, J.; Shotwell, M.S.; Engelman, A.; Aiken, C. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J. Virol. 2013, 87, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Iaco, A.; Luban, J. Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallery, D.L.; Márquez, C.L.; McEwan, W.A.; Dickson, C.F.; Jacques, D.A.; Anandapadamanaban, M.; Bichel, K.; Towers, G.J.; Saiardi, A.; Böcking, T. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife 2018, 7, e35335. [Google Scholar] [CrossRef]
- Dick, R.A.; Zadrozny, K.K.; Xu, C.; Schur, F.K.; Lyddon, T.D.; Ricana, C.L.; Wagner, J.M.; Perilla, J.R.; Ganser-Pornillos, B.K.; Johnson, M.C. Inositol phosphates are assembly co-factors for HIV-1. Nature 2018, 560, 509–512. [Google Scholar] [CrossRef]
- Jacques, D.A.; McEwan, W.A.; Hilditch, L.; Price, A.J.; Towers, G.J.; James, L.C. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 2016, 536, 349–353. [Google Scholar] [CrossRef]
- Xu, C.; Fisher, D.; Rankovic, S.; Li, W.; Dick, R.; Runge, B.; Zadorozhnyi, R.; Ahn, J.; Aiken, C.; Polenova, T. Permeability of the HIV-1 capsid to metabolites modulates viral DNA synthesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Márquez, C.L.; Lau, D.; Walsh, J.; Shah, V.; McGuinness, C.; Wong, A.; Aggarwal, A.; Parker, M.W.; Jacques, D.A.; Turville, S. Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. eLife 2018, 7, e34772. [Google Scholar] [CrossRef]
- Dick, R.A.; Mallery, D.L.; Vogt, V.M.; James, L.C. IP6 regulation of HIV capsid assembly, stability, and uncoating. Viruses 2018, 10, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nunzio, F.; Danckaert, A.; Fricke, T.; Perez, P.; Fernandez, J.; Perret, E.; Roux, P.; Shorte, S.; Charneau, P.; Diaz-Griffero, F. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS ONE 2012, 7, e46037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, E.; Chauhan, R. Host-HIV-1 Interactome: A Quest for Novel Therapeutic Intervention. Cells 2019, 8, 1155. [Google Scholar] [CrossRef] [Green Version]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Panté, N.; Mouland, A.J. HIV-1 remodels the nuclear pore complex. J. Cell Biol. 2011, 193, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Cosnefroy, O.; Murray, P.J.; Bishop, K.N. HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology 2016, 13, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [Green Version]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. bioRxiv 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Stanley, G.J.; Fassati, A.; Hoogenboom, B.W. Atomic force microscopy reveals structural variability amongst nuclear pore complexes. Life Sci. Alliance 2018, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Appen, A.; Beck, M. Structure determination of the nuclear pore complex with three-dimensional cryo electron microscopy. J. Mol. Biol. 2016, 428, 2001–2010. [Google Scholar] [CrossRef] [Green Version]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and assembly of the nuclear pore complex. Annu. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The structure of the nuclear pore complex (an update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [Green Version]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.-Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J. Genome-Scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Yeung, M.L.; Houzet, L.; Yedavalli, V.S.; Jeang, K.-T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009, 284, 19463–19473. [Google Scholar] [CrossRef] [Green Version]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; Kewal Ramani, V.N. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yücel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichel, K.; Price, A.J.; Schaller, T.; Towers, G.J.; Freund, S.M.; James, L.C. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 2013, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meehan, A.M.; Saenz, D.T.; Guevera, R.; Morrison, J.H.; Peretz, M.; Fadel, H.J.; Hamada, M.; Van Deursen, J.; Poeschla, E.M. A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLoS Pathog. 2014, 10, e1003969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.; Walsh, J.C.; Mousapasandi, A.; Ariotti, N.; Shah, V.B.; Turville, S.; Jacques, D.A.; Böcking, T. Self-Assembly of Fluorescent HIV Capsid Spheres for Detection of Capsid Binders. Langmuir 2020, 36, 3624–3632. [Google Scholar] [CrossRef]
- Lin, D.H.; Zimmermann, S.; Stuwe, T.; Stuwe, E.; Hoelz, A. Structural and functional analysis of the C-terminal domain of Nup358/RanBP2. J. Mol. Biol. 2013, 425, 1318–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, J.; Machado, A.K.; Lyonnais, S.; Chamontin, C.; Gärtner, K.; Léger, T.; Henriquet, C.; Garcia, C.; Portilho, D.M.; Pugnière, M. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef]
- Kane, M.; Rebensburg, S.V.; Takata, M.A.; Zang, T.M.; Yamashita, M.; Kvaratskhelia, M.; Bieniasz, P.D. Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. eLife 2018, 7, e35738. [Google Scholar] [CrossRef]
- Takao, D.; Dishinger, J.F.; Kee, H.L.; Pinskey, J.M.; Allen, B.L.; Verhey, K.J. An assay for clogging the ciliary pore complex distinguishes mechanisms of cytosolic and membrane protein entry. Curr. Biol. 2014, 24, 2288–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Börner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, S.; Varadarajan, J.; Ramalho, R.; Aiken, C.; Rousso, I. Reverse transcription mechanically initiates HIV-1 capsid disassembly. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction factors: From intrinsic viral restriction to shaping cellular immunity against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Smaga, S.S.; Xu, C.; Summers, B.J.; Digianantonio, K.M.; Perilla, J.R.; Xiong, Y. MxB restricts HIV-1 by targeting the tri-hexamer interface of the viral capsid. Structure 2019, 27, 1234–1245.e5. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Grütter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent RING assembly on retroviral capsids activates TRIM5 ubiquitination and innate immune signaling. Cell Host Microbe 2018, 24, 761–775.e6. [Google Scholar] [CrossRef] [Green Version]
- Skorupka, K.A.; Roganowicz, M.D.; Christensen, D.E.; Wan, Y.; Pornillos, O.; Ganser-Pornillos, B.K. Hierarchical assembly governs TRIM5α recognition of HIV-1 and retroviral capsids. Sci. Adv. 2019, 5, eaaw3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.; Skorupka, K.A.; Pak, A.J.; Ganser-Pornillos, B.K.; Pornillos, O.; Voth, G.A. TRIM5α self-assembly and compartmentalization of the HIV-1 viral capsid. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Selyutina, A.; Persaud, M.; Simons, L.M.; Bulnes-Ramos, A.; Buffone, C.; Martinez-Lopez, A.; Scoca, V.; Di Nunzio, F.; Hiatt, J.; Marson, A. Cyclophilin A Prevents HIV-1 Restriction in Lymphocytes by Blocking Human TRIM5α Binding to the viral Core. Cell Rep. 2020, 30, 3766–3777.e6. [Google Scholar] [CrossRef]
- Staeheli, P.; Haller, O. Human MX2/MxB: A potent interferon-induced postentry inhibitor of herpesviruses and HIV-1. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, B.; Buffone, C.; Opp, S.; Di Nunzio, F.; Vieira, D.A.D.S.A.; Brandariz-Nuñez, A.; Diaz-Griffero, F. Restriction of HIV-1 requires the N-terminal region of MxB as a capsid-binding motif but not as a nuclear localization signal. J. Virol. 2015, 89, 8599–8610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricke, T.; White, T.E.; Schulte, B.; De Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nuñez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Chen, L.; Zhong, C.; Yu, T.; Ju, Z.; Wang, M.; Xiong, H.; Zeng, Y.; Wang, J.; Hu, H. MxB impedes the NUP358-mediated HIV-1 pre-integration complex nuclear import and viral replication cooperatively with CPSF6. Retrovirology 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Zheng, J.; Yant, S.R.; Ahmadyar, S.; Chan, T.Y.; Chiu, A.; Cihlar, T.; Link, J.O.; Lu, B.; Mwangi, J.; Rowe, W. 539. GS-CA2: A novel, potent, and selective first-in-class inhibitor of HIV-1 capsid function displays nonclinical pharmacokinetics supporting long-acting potential in humans. Open Forum Infect. Dis. 2018, S199. [Google Scholar] [CrossRef]
- Link, J.O.; Rhee, M.S.; Winston, C.T.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 reinforces the HIV-1 capsid to impair reverse transcription-induced uncoating. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring modifications of an HIV-1 capsid inhibitor: Design, synthesis, and mechanism of action. J. Drug Des. Res. 2018, 5, 1070. [Google Scholar] [PubMed]
- Tse, W.; Link, J.; Mulato, A.; Niedziela-Majka, A.; Rowe, W.; Somoza, J.; Villasenor, A.; Yant, S.; Zhang, J.; Zheng, J. Discovery of novel potent HIV capsid inhibitors with long-acting potential. In Proceedings of the Conference on Retroviruses and Opportunistic Infections: 2017, Seattle, WA, USA, 13–16 February 2017; pp. 13–16. [Google Scholar]
- Yant, S.; Mulato, A.; Stepan, G.; Villasenor, A.; Jin, D.; Margot, N.; Ahmadyar, S.; Ram, R.; Somoza, J.; Singer, E. GS-6207, a potent and selective first-in-class long-acting HIV-1 capsid inhibitor. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 4–9 March 2019. [Google Scholar]
- Yant, S.R.; Mulato, A.; Hansen, D.; Winston, C.T.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384. [Google Scholar] [CrossRef]
- Pham, H.T.; Yoo, S.; Mesplède, T. Combination therapies currently under investigation in phase I and phase II clinical trials for HIV-1. Expert Opin. Investig. Drugs 2020, 29, 273–283. [Google Scholar] [CrossRef]
- Daar, E.; McDonald, C.; Crofoot, G.; Ruane, P.; Sinclair, G.; DeJesus, E.; Berhe, M.; Ramgopal, M.; Patel, H.; Liu, Y. Dose-Response relationship of subcutaneous long-acting HIV capsid inhibitor GS-6207. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 8–11 March 2020. [Google Scholar]
- Saito, A.; Ferhadian, D.; Sowd, G.A.; Serrao, E.; Shi, J.; Halambage, U.D.; Teng, S.; Soto, J.; Siddiqui, M.A.; Engelman, A.N. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J. Virol. 2016, 90, 5808–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sönnerborg, A. GS-CA compounds: First-in-class HIV-1 capsid inhibitors covering multiple grounds. Front. Microbiol. 2019, 10, 1227. [Google Scholar] [CrossRef]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D’Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; König, R. Host cell factors in HIV replication: Meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 2018, 7, e39823. [Google Scholar] [CrossRef] [PubMed]
- Koegl, M.; Uetz, P. Improving yeast two-hybrid screening systems. Brief. Funct. Genom. Proteom. 2007, 6, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillen, J.; Nita-Lazar, A. Experimental Analysis of Viral–Host Interactions. Front. Physiol. 2019, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Coombs, K.M. Update on Proteomic approaches to uncovering virus-induced protein alterations and virus–host protein interactions during the progression of viral infection. Expert Rev. Proteom. 2020, 17, 513–532. [Google Scholar] [CrossRef]
- Miteva, Y.V.; Budayeva, H.G.; Cristea, I.M. Proteomics-Based methods for discovery, quantification, and validation of protein–protein interactions. Anal. Chem. 2013, 85, 749–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, T.M.; Diner, B.A.; Cristea, I.M. The impact of mass spectrometry–based proteomics on fundamental discoveries in virology. Annu. Rev. Virol. 2014, 1, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Gulbahce, N.; Cimermancic, P.; Kane, J.; He, N.; Chou, S.; D’Orso, I.; Fernandes, J.; Jang, G.; Frankel, A.D. Purification and characterization of HIV–human protein complexes. Methods 2011, 53, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Milev, M.P.; Ravichandran, M.; Khan, M.F.; Schriemer, D.C.; Mouland, A.J. Characterization of staufen1 ribonucleoproteins by mass spectrometry and biochemical analyses reveal the presence of diverse host proteins associated with human immunodeficiency virus type 1. Front. Microbiol. 2012, 3, 367. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, M.; Hubner, N.C.; Mann, M. High confidence determination of specific protein–protein interactions using quantitative mass spectrometry. Curr. Opin. Biotechnol. 2008, 19, 331–337. [Google Scholar] [CrossRef]
- O’Reilly, F.J.; Rappsilber, J. Cross-Linking mass spectrometry: Methods and applications in structural, molecular and systems biology. Nat. Struct. Mol. Biol. 2018, 25, 1000–1008. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.D.; Cilia, M.; Weisbrod, C.R.; Ju, H.-J.; Eng, J.K.; Gray, S.M.; Bruce, J.E. Cross-Linking measurements of the Potato leafroll virus reveal protein interaction topologies required for virion stability, aphid transmission, and virus-plant interactions. J. Proteome Res. 2012, 11, 2968–2981. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Brown, N.P.; Börner, K.; Schümann, M.; Krause, E.; Kaderali, L.; Müller, G.A.; Kräusslich, H.-G. Proteome analysis of the HIV-1 Gag interactome. Virology 2014, 460, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnaitė, R.; MacNeill, S.A. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 2016, 16, 2503–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Birendra, K.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Sage, V.; Cinti, A.; Valiente-Echeverría, F.; Mouland, A.J. Proteomic analysis of HIV-1 Gag interacting partners using proximity-dependent biotinylation. Virol. J. 2015, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Ako-Adjei, D.; Fu, W.; Wallin, C.; Katz, K.S.; Song, G.; Darji, D.; Brister, J.R.; Ptak, R.G.; Pruitt, K.D. HIV-1, human interaction database: Current status and new features. Nucleic Acids Res. 2015, 43, D566–D570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Tekir, S.D.; Çakır, T.; Sayılırbaş, A.S.; Çelik, E.; Özcan, S.; Çevik, İ.; Özçelik, A.S.; Özgür, A.; Sevilgen, F.E.; Ülgen, K.Ö. PHISTO: Pathogen-Host Interaction Search Tool. New Biotechnol. 2012, S151. [Google Scholar] [CrossRef]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, H.V.; Doncheva, N.T.; Szklarczyk, D.; Von Mering, C.; Jensen, L.J. Viruses. STRING: A virus-host protein-protein interaction database. Viruses 2018, 10, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderone, A.; Licata, L.; Cesareni, G. VirusMentha: A new resource for virus-host protein interactions. Nucleic Acids Res. 2015, 43, D588–D592. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approaches | Advantages | Limitations | |
|---|---|---|---|
| Genomic approaches | RNAi |
|
|
| CRISPR |
|
| |
| Proteomic approaches | Yeast two-hybrid (Y2H) |
|
|
| AP-MS |
|
| |
| AP-MS coupled with SILAC |
|
| |
| AP-MS coupled with chemical crossing-linking |
|
| |
| Proximity-dependent labeling (PDL) |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, S.; Torbett, B.E. Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics. Viruses 2021, 13, 417. https://doi.org/10.3390/v13030417
Zhuang S, Torbett BE. Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics. Viruses. 2021; 13(3):417. https://doi.org/10.3390/v13030417
Chicago/Turabian StyleZhuang, Shentian, and Bruce E. Torbett. 2021. "Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics" Viruses 13, no. 3: 417. https://doi.org/10.3390/v13030417
APA StyleZhuang, S., & Torbett, B. E. (2021). Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics. Viruses, 13(3), 417. https://doi.org/10.3390/v13030417