
HCMV Antivirals and Strategies to Target the Latent Reservoir




{kind=link}
Abstract
:1. Introduction
2. Current HCMV Antivirals
2.1. Ganciclovir
2.2. Foscarnet
2.3. Cidofovir
2.4. Letermovir
2.5. Maribavir
2.6. Antibody Therapies
3. Strategies for Targeting the Latent HCMV Reservoir
3.1. Potential ‘Shock and Kill’ Treatments to Purge the Latent HCMV Reservoir
3.2. Eradicating the Latent Reservoir by Targeting Viral Genes Expressed during Latency
3.2.1. Vincristine and UL138
3.2.2. F49A Fusion Toxin Protein to Target US28
3.2.3. US28 Inhibitors as a Novel Method for ‘Shock and Kill’
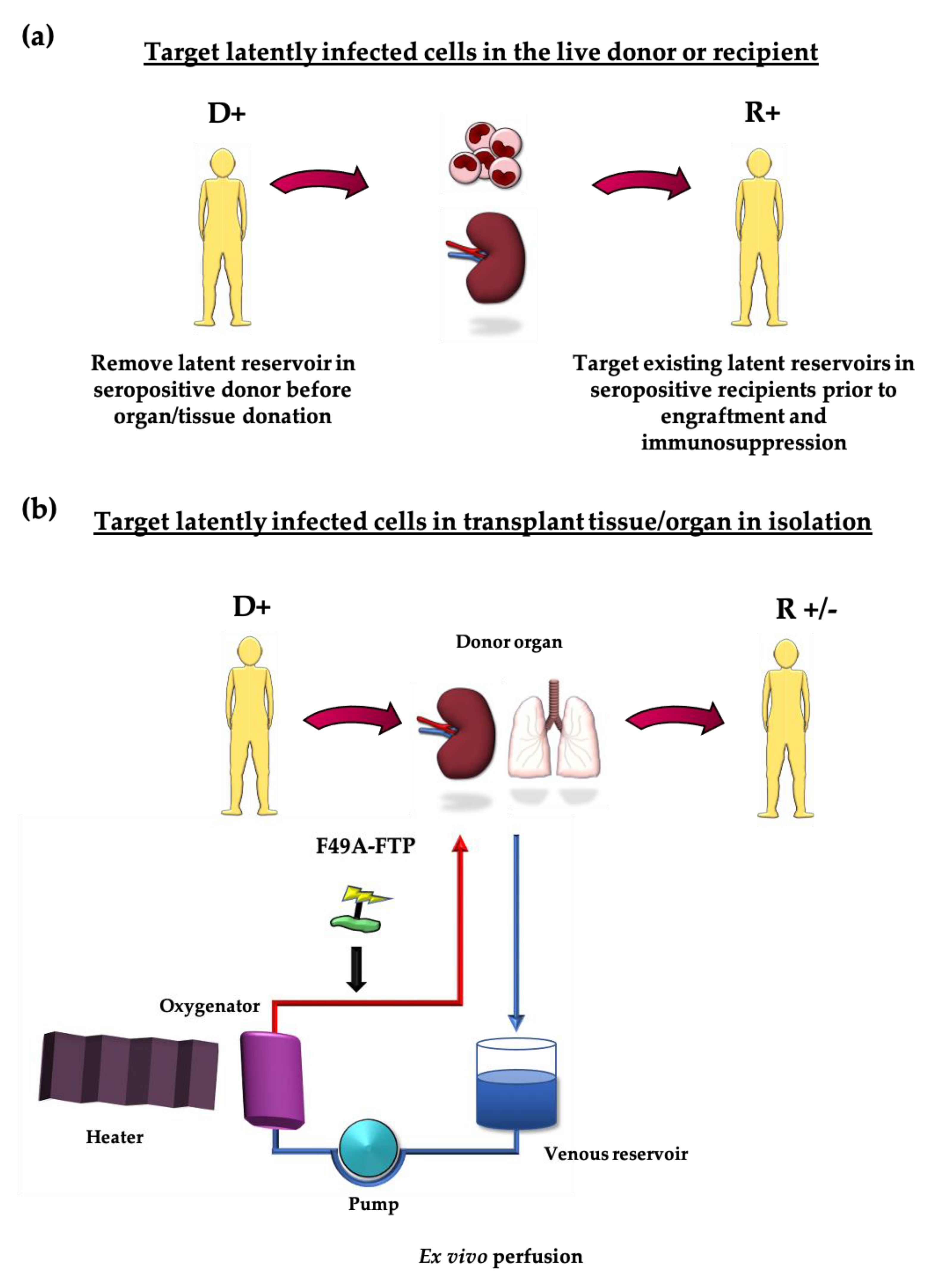
4. Targeting the Latent Reservoir in the Transplant Scenario
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of Cytomegalovirus Seroprevalence and Demographic Characteristics Associated with Infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Zhao, X.-Y. Human Cytomegalovirus Primary Infection and Reactivation: Insights from Virion-Carried Molecules. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Dell’Oste, V.; Biolatti, M.; Galitska, G.; Griffante, G.; Gugliesi, F.; Pasquero, S.; Zingoni, A.; Cerboni, C.; De Andrea, M. Tuning the Orchestra: HCMV vs. Innate Immunity. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Picarda, G.; Benedict, C.A. Cytomegalovirus: Shape-Shifting the Immune System. J. Immunol. 2018, 200, 3881–3889. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human Cytomegalovirus Immunity and Immune Evasion. Virus Res. 2011, 157, 151–160. [Google Scholar] [CrossRef]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus Remains Latent in a Common Precursor of Dendritic and Myeloid Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of Endogenous Human Cytomegalovirus in CD34+ Bone Marrow Progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes Are a Major Site of Persistence of Human Cytomegalovirus in Peripheral Blood Mononuclear Cells. J. Gen. Virol. 1991, 72 Pt 9, 2059–2064. [Google Scholar] [CrossRef]
- Griffiths, P.; Baraniak, I.; Reeves, M. The Pathogenesis of Human Cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Lim, Y.; Lyall, H. Congenital Cytomegalovirus—Who, When, What-with and Why to Treat? J. Infect. 2017, 74, S89–S94. [Google Scholar] [CrossRef]
- Grosse, S.D.; Ross, D.S.; Dollard, S.C. Congenital Cytomegalovirus (CMV) Infection as a Cause of Permanent Bilateral Hearing Loss: A Quantitative Assessment. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2008, 41, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New Estimates of the Prevalence of Neurological and Sensory Sequelae and Mortality Associated with Congenital Cytomegalovirus Infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, X.; Bialek, S.; Cannon, M.J. Attribution of Congenital Cytomegalovirus Infection to Primary versus Non-Primary Maternal Infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2011, 52, e11–e13. [Google Scholar] [CrossRef]
- de Vries, J.J.C.; van Zwet, E.W.; Dekker, F.W.; Kroes, A.C.M.; Verkerk, P.H.; Vossen, A.C.T.M. The Apparent Paradox of Maternal Seropositivity as a Risk Factor for Congenital Cytomegalovirus Infection: A Population-Based Prediction Model. Rev. Med. Virol. 2013, 23, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Leruez-Ville, M.; Magny, J.-F.; Couderc, S.; Pichon, C.; Parodi, M.; Bussières, L.; Guilleminot, T.; Ghout, I.; Ville, Y. Risk Factors for Congenital Cytomegalovirus Infection Following Primary and Nonprimary Maternal Infection: A Prospective Neonatal Screening Study Using Polymerase Chain Reaction in Saliva. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017, 65, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.S.; Alford, C.A.; Reynolds, D.W.; Stagno, S.; Pass, R.F. Molecular Epidemiology of Cytomegalovirus Infections in Women and Their Infants. N. Engl. J. Med. 1980, 303, 958–962. [Google Scholar] [CrossRef]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine Transmission of Cytomegalovirus to Infants of Women with Preconceptional Immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar] [CrossRef]
- Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Moura Britto, R.M.; de Lima Isaac, M.; de Carvalho e Oliveira, P.F.; Boppana, S.; Britt, W.J. Birth Prevalence and Natural History of Congenital Cytomegalovirus (CMV) Infection in a Highly Seroimmune Population. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 49, 522–528. [Google Scholar] [CrossRef]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus Cell Tropism. In Human Cytomegalovirus; Shenk, T.E., Stinski, M.F., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 63–83. ISBN 978-3-540-77349-8. [Google Scholar]
- Britt, W. Virus entry into host, establishment of infection, spread in host, mechanisms of tissue damage. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- Atabani, S.F.; Smith, C.; Atkinson, C.; Aldridge, R.W.; Rodriguez-Perálvarez, M.; Rolando, N.; Harber, M.; Jones, G.; O’Riordan, A.; Burroughs, A.K.; et al. Cytomegalovirus Replication Kinetics in Solid Organ Transplant Recipients Managed by Preemptive Therapy. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2012, 12, 2457–2464. [Google Scholar] [CrossRef]
- Kotton, C.N. Management of Cytomegalovirus Infection in Solid Organ Transplantation. Nat. Rev. Nephrol. 2010, 6, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Grundy, J.E.; Lui, S.F.; Super, M.; Berry, N.J.; Sweny, P.; Fernando, O.N.; Moorhead, J.; Griffiths, P.D. Symptomatic Cytomegalovirus Infection in Seropositive Kidney Recipients: Reinfection with Donor Virus Rather than Reactivation of Recipient Virus. Lancet Lond. Engl. 1988, 2, 132–135. [Google Scholar] [CrossRef]
- George, B.; Pati, N.; Gilroy, N.; Ratnamohan, M.; Huang, G.; Kerridge, I.; Hertzberg, M.; Gottlieb, D.; Bradstock, K. Pre-Transplant Cytomegalovirus (CMV) Serostatus Remains the Most Important Determinant of CMV Reactivation after Allogeneic Hematopoietic Stem Cell Transplantation in the Era of Surveillance and Preemptive Therapy. Transpl. Infect. Dis. 2010, 12, 322–329. [Google Scholar] [CrossRef]
- Krishna, B.A.; Wills, M.R.; Sinclair, J.H. Advances in the Treatment of Cytomegalovirus. Br. Med. Bull. 2019, 131, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The Immunology of Human Cytomegalovirus Latency: Could Latent Infection Be Cleared by Novel Immunotherapeutic Strategies? Cell. Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Froberg, M.K. CMV Escapes! Ann. Clin. Lab. Sci. 2004, 34, 123–130. [Google Scholar]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The Story of Human Cytomegalovirus and Cancer: Increasing Evidence and Open Questions. Neoplasia 2009, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleiss, M.R. Cytomegalovirus Vaccine Development. In Human Cytomegalovirus; Shenk, T.E., Stinski, M.F., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/ Heidelberg, Germany, 2008; pp. 361–382. ISBN 978-3-540-77349-8. [Google Scholar]
- Martin, J.C.; Dvorak, C.A.; Smee, D.F.; Matthews, T.R.; Verheyden, J.P.H. 9-(1,3-Dihydroxy-2-Propoxymethyl)Guanine: A New Potent and Selective Antiherpes Agent. J. Med. Chem. 1983, 26, 759–761. [Google Scholar] [CrossRef]
- Littler, E.; Stuart, A.D.; Chee, M.S. Human Cytomegalovirus UL97 Open Reading Frame Encodes a Protein That Phosphorylates the Antiviral Nucleoside Analogue Ganciclovir. Nature 1992, 358, 160–162. [Google Scholar] [CrossRef]
- Talarico, C.L.; Burnette, T.C.; Miller, W.H.; Smith, S.L.; Davis, M.G.; Stanat, S.C.; Ng, T.I.; He, Z.; Coen, D.M.; Roizman, B.; et al. Acyclovir Is Phosphorylated by the Human Cytomegalovirus UL97 Protein. Antimicrob. Agents Chemother. 1999, 43, 1941–1946. [Google Scholar] [CrossRef] [Green Version]
- Braakman, E.; Vogels, R.; Martens, A.C.M.; Vermeulen, J.; Bron, M.; Hoogerbrugge, P.M.; Valerio, D.; Hagenbeek, A. Ganciclovir-Mediated in Vivo Elimination of Myeloid Leukemic Cells Expressing the HSVtk Gene Induces HSVtk Loss Variants. Gene Ther. 1999, 6, 1139–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, V.R.; Smee, D.F.; Chernow, M.; Boehme, R.; Matthews, T.R. Activity of 9-(1,3-Dihydroxy-2-Propoxymethyl)Guanine Compared with That of Acyclovir against Human, Monkey, and Rodent Cytomegaloviruses. Antimicrob. Agents Chemother. 1985, 28, 240–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mar, E.C.; Chiou, J.F.; Cheng, Y.C.; Huang, E.S. Inhibition of Cellular DNA Polymerase Alpha and Human Cytomegalovirus-Induced DNA Polymerase by the Triphosphates of 9-(2-Hydroxyethoxymethyl)Guanine and 9-(1,3-Dihydroxy-2-Propoxymethyl)Guanine. J. Virol. 1985, 53, 776–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Beardsley, G.P.; Coen, D.M. Mechanism of Ganciclovir-Induced Chain Termination Revealed by Resistant Viral Polymerase Mutants with Reduced Exonuclease Activity. Proc. Natl. Acad. Sci. USA 2014, 111, 17462–17467. [Google Scholar] [CrossRef] [Green Version]
- Biron, K.K.; Stanat, S.C.; Sorrell, J.B.; Fyfe, J.A.; Keller, P.M.; Lambe, C.U.; Nelson, D.J. Metabolic Activation of the Nucleoside Analog 9-[(2-Hydroxy-1-(Hydroxymethyl)Ethoxy]Methyl)Guanine in Human Diploid Fibroblasts Infected with Human Cytomegalovirus. Proc. Natl. Acad. Sci. USA 1985, 82, 2473–2477. [Google Scholar] [CrossRef] [Green Version]
- Fortún Abete, J.; Martín-Dávila, P.; Moreno, S.; Quijano, Y.; de Vicente, E.; Pou, L. Pharmacokinetics of Oral Valganciclovir and Intravenous Ganciclovir Administered to Prevent Cytomegalovirus Disease in an Adult Patient Receiving Small-Intestine Transplantation. Antimicrob. Agents Chemother. 2004, 48, 2782–2783. [Google Scholar] [CrossRef] [Green Version]
- Pescovitz, M.D.; Rabkin, J.; Merion, R.M.; Paya, C.V.; Pirsch, J.; Freeman, R.B.; O’Grady, J.; Robinson, C.; To, Z.; Wren, K.; et al. Valganciclovir Results in Improved Oral Absorption of Ganciclovir in Liver Transplant Recipients. Antimicrob. Agents Chemother. 2000, 44, 2811–2815. [Google Scholar] [CrossRef] [Green Version]
- Steininger, C. Novel Therapies for Cytomegalovirus Disease. Recent Patents Anti Infect. Drug Discov. 2007, 2, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Limaye, A.P. Ganciclovir-Resistant Cytomegalovirus in Organ Transplant Recipients. Clin. Infect. Dis. 2002, 35, 866–872. [Google Scholar] [CrossRef]
- Mumtaz, K.; Faisal, N.; Husain, S.; Morillo, A.; Renner, E.L.; Shah, P.S. Universal Prophylaxis or Preemptive Strategy for Cytomegalovirus Disease After Liver Transplantation: A Systematic Review and Meta-Analysis. Am. J. Transplant. 2015, 15, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Humar, A.; Lebranchu, Y.; Vincenti, F.; Blumberg, E.A.; Punch, J.D.; Limaye, A.P.; Abramowicz, D.; Jardine, A.G.; Voulgari, A.T.; Ives, J.; et al. The Efficacy and Safety of 200 Days Valganciclovir Cytomegalovirus Prophylaxis in High-Risk Kidney Transplant Recipients. Am. J. Transplant. 2010, 10, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Battiwalla, M.; Wu, Y.; Bajwa, R.P.S.; Radovic, M.; Almyroudis, N.G.; Segal, B.H.; Wallace, P.K.; Nakamura, R.; Padmanabhan, S.; Hahn, T.; et al. Ganciclovir Inhibits Lymphocyte Proliferation by Impairing DNA Synthesis. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2007, 13, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Venton, G.; Crocchiolo, R.; Fürst, S.; Granata, A.; Oudin, C.; Faucher, C.; Coso, D.; Bouabdallah, R.; Berger, P.; Vey, N.; et al. Risk Factors of Ganciclovir-Related Neutropenia after Allogeneic Stem Cell Transplantation: A Retrospective Monocentre Study on 547 Patients. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Crumpacker, C.S. Mechanism of Action of Foscarnet against Viral Polymerases. Am. J. Med. 1992, 92, S3–S7. [Google Scholar] [CrossRef]
- Eriksson, B.; Oberg, B.; Wahren, B. Pyrophosphate Analogues as Inhibitors of DNA Polymerases of Cytomegalovirus, Herpes Simplex Virus and Cellular Origin. Biochim. Biophys. Acta 1982, 696, 115–123. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Bryson, H.M. Foscarnet. A Reappraisal of Its Antiviral Activity, Pharmacokinetic Properties and Therapeutic Use in Immunocompromised Patients with Viral Infections. Drugs 1994, 48, 199–226. [Google Scholar] [CrossRef] [PubMed]
- Minor, J.R.; Baltz, J.K. Foscarnet Sodium. DICP Ann. Pharmacother. 1991, 25, 41–47. [Google Scholar] [CrossRef]
- Cihlar, T.; Chen, M.S. Identification of Enzymes Catalyzing Two-Step Phosphorylation of Cidofovir and the Effect of Cytomegalovirus Infection on Their Activities in Host Cells. Mol. Pharmacol. 1996, 50, 1502–1510. [Google Scholar]
- Xiong, X.; Smith, J.L.; Chen, M.S. Effect of Incorporation of Cidofovir into DNA by Human Cytomegalovirus DNA Polymerase on DNA Elongation. Antimicrob. Agents Chemother. 1997, 41, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, A.B.; Ribeiro, J.; Boutolleau, D.; Sousa, H. Human Cytomegalovirus Antiviral Drug Resistance in Hematopoietic Stem Cell Transplantation: Current State of the Art. Rev. Med. Virol. 2016, 26, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Acosta, E.; Bowlin, T.; Brooks, J.; Chiang, L.; Hussein, I.; Kimberlin, D.; Kauvar, L.M.; Leavitt, R.; Prichard, M.; Whitley, R. Advances in the Development of Therapeutics for Cytomegalovirus Infections. J. Infect. Dis. 2020, 221, S32–S44. [Google Scholar] [CrossRef]
- Tippin, T.K.; Morrison, M.E.; Brundage, T.M.; Momméja-Marin, H. Brincidofovir Is Not a Substrate for the Human Organic Anion Transporter 1: A Mechanistic Explanation for the Lack of Nephrotoxicity Observed in Clinical Studies. Ther. Drug Monit. 2016, 38, 777–786. [Google Scholar] [CrossRef] [Green Version]
- El Chaer, F.; Shah, D.P.; Chemaly, R.F. How I Treat Resistant Cytomegalovirus Infection in Hematopoietic Cell Transplantation Recipients. Blood 2016, 128, 2624–2636. [Google Scholar] [CrossRef] [Green Version]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The Novel Anticytomegalovirus Compound AIC246 (Letermovir) Inhibits Human Cytomegalovirus Replication through a Specific Antiviral Mechanism That Involves the Viral Terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Shigle, T.L.; Handy, V.W.; Chemaly, R.F. Letermovir and Its Role in the Prevention of Cytomegalovirus Infection in Seropositive Patients Receiving an Allogeneic Hematopoietic Cell Transplant. Ther. Adv. Hematol. 2020, 11. [Google Scholar] [CrossRef]
- Kim, E.S. Letermovir: First Global Approval. Drugs 2018, 78, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Maertens, J.; Cordonnier, C.; Jaksch, P.; Poiré, X.; Uknis, M.; Wu, J.; Wijatyk, A.; Saliba, F.; Witzke, O.; Villano, S. Maribavir for Preemptive Treatment of Cytomegalovirus Reactivation. N. Engl. J. Med. 2019, 381, 1136–1147. [Google Scholar] [CrossRef]
- Drew, W.L.; Miner, R.C.; Marousek, G.I.; Chou, S. Maribavir Sensitivity of Cytomegalovirus Isolates Resistant to Ganciclovir, Cidofovir or Foscarnet. J. Clin. Virol. 2006, 37, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Marousek, G.I. Maribavir Antagonizes the Antiviral Action of Ganciclovir on Human Cytomegalovirus. Antimicrob. Agents Chemother. 2006, 50, 3470–3472. [Google Scholar] [CrossRef] [Green Version]
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. National Vaccine Advisory Committee Vaccine Development to Prevent Cytomegalovirus Disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2004, 39, 233–239. [Google Scholar] [CrossRef]
- Gerna, G.; Sarasini, A.; Patrone, M.; Percivalle, E.; Fiorina, L.; Campanini, G.; Gallina, A.; Baldanti, F.; Revello, M.G. Human Cytomegalovirus Serum Neutralizing Antibodies Block Virus Infection of Endothelial/Epithelial Cells, but Not Fibroblasts, Early during Primary Infection. J. Gen. Virol. 2008, 89, 853–865. [Google Scholar] [CrossRef]
- Kauvar, L.M.; Liu, K.; Park, M.; DeChene, N.; Stephenson, R.; Tenorio, E.; Ellsworth, S.L.; Tabata, T.; Petitt, M.; Tsuge, M.; et al. A High-Affinity Native Human Antibody Neutralizes Human Cytomegalovirus Infection of Diverse Cell Types. Antimicrob. Agents Chemother. 2015, 59, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Bonaros, N.; Mayer, B.; Schachner, T.; Laufer, G.; Kocher, A. CMV-Hyperimmune Globulin for Preventing Cytomegalovirus Infection and Disease in Solid Organ Transplant Recipients: A Meta-Analysis. Clin. Transplant. 2008, 22, 89–97. [Google Scholar] [CrossRef]
- Ishida, J.H.; Patel, A.; Mehta, A.K.; Gatault, P.; McBride, J.M.; Burgess, T.; Derby, M.A.; Snydman, D.R.; Emu, B.; Feierbach, B.; et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of RG7667, a Combination Monoclonal Antibody, for Prevention of Cytomegalovirus Infection in High-Risk Kidney Transplant Recipients. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; Coleman, H.; Chadderton, J.; Goddard, M.; Sissons, J.G.P.; Sinclair, J.H. Vascular Endothelial and Smooth Muscle Cells Are Unlikely to Be Major Sites of Latency of Human Cytomegalovirus in Vivo. J. Gen. Virol. 2004, 85, 3337–3341. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium: I. Structure, Function, and Mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koffron, A.J.; Hummel, M.; Patterson, B.K.; Yan, S.; Kaufman, D.B.; Fryer, J.P.; Stuart, F.P.; Abecassis, M.I. Cellular Localization of Latent Murine Cytomegalovirus. J. Virol. 1998, 72, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Seckert, C.K.; Renzaho, A.; Tervo, H.-M.; Krause, C.; Deegen, P.; Kühnapfel, B.; Reddehase, M.J.; Grzimek, N.K.A. Liver Sinusoidal Endothelial Cells Are a Site of Murine Cytomegalovirus Latency and Reactivation. J. Virol. 2009, 83, 8869–8884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Žugich, J.; Goodrum, F. Transcriptome-Wide Characterization of Human Cytomegalovirus in Natural Infection and Experimental Latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human Cytomegalovirus Gene Expression during Infection of Primary Hematopoietic Progenitor Cells: A Model for Latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Mocarski, E.S. Cytomegalovirus Latency and Latency-Specific Transcription in Hematopoietic Progenitors. Scand. J. Infect. Dis. Suppl. 1995, 99, 63–67. [Google Scholar] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Analysis of Latent Viral Gene Expression in Natural and Experimental Latency Models of Human Cytomegalovirus and Its Correlation with Histone Modifications at a Latent Promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. mBio 2018, 9, e00013-18. [Google Scholar] [CrossRef] [Green Version]
- Elder, E.; Sinclair, J. HCMV Latency: What Regulates the Regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, A.L.; O’Connor, C.M. Regulation of the MIE Locus During HCMV Latency and Reactivation. Pathogens 2020, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B. Cell Signaling and Cytomegalovirus Reactivation: What Do Src Family Kinases Have to Do with It? Biochem. Soc. Trans. 2020, 48, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Vogel, J.L.; Arbuckle, J.H.; Rai, G.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; Kristie, T.M. Targeting the JMJD2 Histone Demethylases to Epigenetically Control Herpesvirus Infection and Reactivation from Latency. Sci. Transl. Med. 2013, 5, 167ra5. [Google Scholar] [CrossRef] [Green Version]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of Viral Infections by Epigenetic-Targeted Therapy. Clin. Epigenetics 2019, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Dağ, F.; Dölken, L.; Holzki, J.; Drabig, A.; Weingärtner, A.; Schwerk, J.; Lienenklaus, S.; Conte, I.; Geffers, R.; Davenport, C.; et al. Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency. PLoS Pathog. 2014, 10, e1003962. [Google Scholar] [CrossRef] [Green Version]
- Presti, R.M.; Pollock, J.L.; Dal Canto, A.J.; O’Guin, A.K.; Virgin, H.W. Interferon Gamma Regulates Acute and Latent Murine Cytomegalovirus Infection and Chronic Disease of the Great Vessels. J. Exp. Med. 1998, 188, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, L.; Du, L.; Poulter, M.; Choi, S.; McIntosh, M.; Reeves, M.B. Src Family Kinase Activity Drives Cytomegalovirus Reactivation by Recruiting MOZ Histone Acetyltransferase Activity to the Viral Promoter. J. Biol. Chem. 2019, 294, 12901–12910. [Google Scholar] [CrossRef] [Green Version]
- Kew, V.G.; Yuan, J.; Meier, J.; Reeves, M.B. Mitogen and Stress Activated Kinases Act Co-Operatively with CREB during the Induction of Human Cytomegalovirus Immediate-Early Gene Expression from Latency. PLoS Pathog. 2014, 10, e1004195. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Wass, A.B.; Dooley, A.L.; O’Connor, C.M. CMV-Encoded GPCR PUL33 Activates CREB and Facilitates Its Recruitment to the MIE Locus for Efficient Viral Reactivation. J. Cell Sci. 2021, 134, jcs254268. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Ruggiero, A.; Paxton, W.A.; Pollakis, G. Measuring the Success of HIV-1 Cure Strategies. Front. Cell. Infect. Microbiol. 2020, 10, 134. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “Shock and Kill” Therapy: In Need of Revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. An in Vitro Model for the Regulation of Human Cytomegalovirus Latency and Reactivation in Dendritic Cells by Chromatin Remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and Trans Acting Factors Involved in Human Cytomegalovirus Experimental and Natural Latent Infection of CD14 (+) Monocytes and CD34 (+) Cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J. Chromatin Structure Regulates Human Cytomegalovirus Gene Expression during Latency, Reactivation and Lytic Infection. Biochim. Biophys. Acta 2010, 1799, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. Latency, Chromatin Remodeling, and Reactivation of Human Cytomegalovirus in the Dendritic Cells of Healthy Carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humby, M.S.; O’Connor, C.M. Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J. Virol. 2016, 90, 2959–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.-H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. J. Virol. 2016, 90, 9483–9494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauwel, B.; Jang, S.M.; Cassano, M.; Kapopoulou, A.; Barde, I.; Trono, D. Release of Human Cytomegalovirus from Latency by a KAP1/TRIM28 Phosphorylation Switch. eLife 2015, 4, e06068. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.; Sinclair, J. Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter. Viruses 2013, 5, 1395–1413. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Lau, B.; Jackson, S.E.; Wills, M.R.; Sinclair, J.H.; Poole, E. Transient Activation of Human Cytomegalovirus Lytic Gene Expression during Latency Allows Cytotoxic T Cell Killing of Latently Infected Cells. Sci. Rep. 2016, 6, srep24674. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly Targeted Human Cytomegalovirus-Specific CD4+ and CD8+ T Cells Dominate the Memory Compartments of Exposed Subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Cobbold, M.; Keenan, R.; Moss, P.A.H. Comparative Analysis of CD8+ T Cell Responses against Human Cytomegalovirus Proteins Pp65 and Immediate Early 1 Shows Similarities in Precursor Frequency, Oligoclonality, and Phenotype. J. Infect. Dis. 2002, 185, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain Proteins Regulate Human Cytomegalovirus Latency and Reactivation Allowing Epigenetic Therapeutic Intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2023025118. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Sinclair, J.H.; Wills, M.R. Bromodomain Inhibitors as Therapeutics for Herpesvirus-Related Disease: All BETs Are Off? Front. Cell. Infect. Microbiol. 2020, 10, 329. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and Extra-Terminal (BET) Bromodomain Inhibition Activate Transcription via Transient Release of Positive Transcription Elongation Factor b (P-TEFb) from 7SK Small Nuclear Ribonucleoprotein *. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Dunn, R.; Turner, A.-M.W.; Jean Beltran, P.M.; Arbuckle, J.H.; Budayeva, H.G.; Cristea, I.M.; Kristie, T.M. Transcriptional Elongation of HSV Immediate Early Genes by the Super Elongation Complex Drives Lytic Infection and Reactivation from Latency. Cell Host Microbe 2017, 21, 507–517.e5. [Google Scholar] [CrossRef] [Green Version]
- Britt, W.J.; Mach, M. Human Cytomegalovirus Glycoproteins. Intervirology 1996, 39, 401–412. [Google Scholar] [CrossRef]
- Wills, M.R.; Carmichael, A.J.; Mynard, K.; Jin, X.; Weekes, M.P.; Plachter, B.; Sissons, J.G. The Human Cytotoxic T-Lymphocyte (CTL) Response to Cytomegalovirus Is Dominated by Structural Protein Pp65: Frequency, Specificity, and T-Cell Receptor Usage of Pp65-Specific CTL. J. Virol. 1996, 70, 7569–7579. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Swaminathan, S.; Schroeder, M.W.; Kim, J.Y.; Terhune, S.S.; Hummel, M. Tumor Necrosis Factor Alpha Induces Reactivation of Human Cytomegalovirus Independently of Myeloid Cell Differentiation Following Posttranscriptional Establishment of Latency. mBio 2018, 9, e01560-18. [Google Scholar] [CrossRef] [Green Version]
- Montag, C.; Wagner, J.A.; Gruska, I.; Vetter, B.; Wiebusch, L.; Hagemeier, C. The Latency-Associated UL138 Gene Product of Human Cytomegalovirus Sensitizes Cells to Tumor Necrosis Factor Alpha (TNF-Alpha) Signaling by Upregulating TNF-Alpha Receptor 1 Cell Surface Expression. J. Virol. 2011, 85, 11409–11421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korngold, R.; Marini, J.C.; de Baca, M.E.; Murphy, G.F.; Giles-Komar, J. Role of Tumor Necrosis Factor-Alpha in Graft-versus-Host Disease and Graft-versus-Leukemia Responses. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2003, 9, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ning, H.-M.; Li, P.-L.; Yan, H.-M.; Han, D.-M.; Zheng, X.-L.; Liu, J.; Zhu, L.; Xue, M.; Mao, N.; et al. Tumor Necrosis Factor α in AGVHD Patients Contributed to the Impairment of Recipient Bone Marrow MSC Stemness and Deficiency of Their Hematopoiesis-Promotion Capacity. Stem Cell Res. Ther. 2020, 11, 119. [Google Scholar] [CrossRef]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F. Opposing Regulation of the EGF Receptor: A Molecular Switch Controlling Cytomegalovirus Latency and Replication. PLoS Pathog. 2016, 12, e1005655. [Google Scholar] [CrossRef] [Green Version]
- Buehler, J.; Carpenter, E.; Zeltzer, S.; Igarashi, S.; Rak, M.; Mikell, I.; Nelson, J.A.; Goodrum, F. Host Signaling and EGR1 Transcriptional Control of Human Cytomegalovirus Replication and Latency. PLoS Pathog. 2019, 15, e1008037. [Google Scholar] [CrossRef]
- Mlera, L.; Moy, M.; Maness, K.; Tran, L.N.; Goodrum, F.D. The Role of the Human Cytomegalovirus UL133-UL138 Gene Locus in Latency and Reactivation. Viruses 2020, 12, 714. [Google Scholar] [CrossRef]
- Young, L.C.; Campling, B.G.; Cole, S.P.C.; Deeley, R.G.; Gerlach, J.H. Multidrug Resistance Proteins MRP3, MRP1, and MRP2 in Lung Cancer: Correlation of Protein Levels with Drug Response and Messenger RNA Levels. Clin. Cancer Res. 2001, 7, 1798–1804. [Google Scholar]
- Weekes, M.P.; Tan, S.Y.L.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-Associated Degradation of the MRP1 Drug Transporter during Latent Human Cytomegalovirus Infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Wills, M.; Sinclair, J. Human Cytomegalovirus Latency: Targeting Differences in the Latently Infected Cell with a View to Clearing Latent Infection. Available online: https://www.hindawi.com/journals/njos/2014/313761/ (accessed on 24 July 2017).
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the Latent Cytomegalovirus Reservoir with an Antiviral Fusion Toxin Protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, B.A.; Miller, W.E.; O’Connor, C.M. US28: HCMV’s Swiss Army Knife. Viruses 2018, 10, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, E.; Krishna, B.; Williamson, J.; Aslam, Y.; Farahi, N.; Wood, A.; Romashova, V.; Roche, K.; Murphy, E.; Chilvers, E.; et al. Monocytes Latently Infected with Human Cytomegalovirus Evade Neutrophil Killing. iScience 2019, 12, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, K.; Jeppesen, M.G.; Malmgaard-Clausen, M.; Krzywkowski, K.; Dulal, K.; Cheng, T.; Hjortø, G.M.; Larsen, O.; Burg, J.S.; Jarvis, M.A.; et al. Rationally Designed Chemokine-Based Toxin Targeting the Viral G Protein-Coupled Receptor US28 Potently Inhibits Cytomegalovirus Infection in Vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 8427–8432. [Google Scholar] [CrossRef] [Green Version]
- Casarosa, P.; Menge, W.M.; Minisini, R.; Otto, C.; van Heteren, J.; Jongejan, A.; Timmerman, H.; Moepps, B.; Kirchhoff, F.; Mertens, T.; et al. Identification of the First Nonpeptidergic Inverse Agonist for a Constitutively Active Viral-Encoded G Protein-Coupled Receptor. J. Biol. Chem. 2003, 278, 5172–5178. [Google Scholar] [CrossRef] [Green Version]
- Groof, T.W.M.D.; Elder, E.G.; Heukers, R.; Lim, E.Y.; Wills, M.; Sinclair, J.H.; Smit, M.J. Targeting the Latent Human Cytomegalovirus Reservoir with Virus Specific Nanobodies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ku, T.J.Y.; Ribeiro, R.V.P.; Ferreira, V.H.; Galasso, M.; Keshavjee, S.; Kumar, D.; Cypel, M.; Humar, A. Ex-Vivo Delivery of Monoclonal Antibody (Rituximab) to Treat Human Donor Lungs Prior to Transplantation. EBioMedicine 2020, 60, 102994. [Google Scholar] [CrossRef] [PubMed]
- Galasso, M.; Feld, J.J.; Watanabe, Y.; Pipkin, M.; Summers, C.; Ali, A.; Qaqish, R.; Chen, M.; Ribeiro, R.V.P.; Ramadan, K.; et al. Inactivating Hepatitis C Virus in Donor Lungs Using Light Therapies during Normothermic Ex Vivo Lung Perfusion. Nat. Commun. 2019, 10, 481. [Google Scholar] [CrossRef]
- Haese, N.N.; Burg, J.M.; Andoh, T.F.; Jones, I.K.A.; Kreklywich, C.N.; Smith, P.P.; Orloff, S.L.; Streblow, D.N. Macrophage Depletion of CMV Latently Infected Donor Hearts Ameliorates Recipient Accelerated Chronic Rejection. Transpl. Infect. Dis. 2020, 23, e13514. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.V.; Ku, T.; Ferreira, V.H.; Galasso, M.; Moshkelgosha, S.; Michaelsen, V.; Wang, A.; Ali, A.; Ramadan, K.; Gomes, B.M.; et al. Targeting Latent Human Cytomegalovirus (CMV) with a Novel Fusion Toxin Protein during Ex Vivo Lung Perfusion. J. Heart Lung Transplant. 2020, 39, S83. [Google Scholar] [CrossRef]
- Cypel, M. Ex Vivo Lung Perfusion (EVLP). Curr. Respir. Care Rep. 2013, 2, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Cypel, M.; Keshavjee, S. Ex Vivo Lung Perfusion. Oper. Tech. Thorac. Cardiovasc. Surg. 2014, 19, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Cypel, M.; Liu, M.; Rubacha, M.; Yeung, J.C.; Hirayama, S.; Anraku, M.; Sato, M.; Medin, J.; Davidson, B.L.; de Perrot, M.; et al. Functional Repair of Human Donor Lungs by IL-10 Gene Therapy. Sci. Transl. Med. 2009, 1, 4ra9. [Google Scholar] [CrossRef]
- Slobedman, B.; Mocarski, E.S. Quantitative Analysis of Latent Human Cytomegalovirus. J. Virol. 1999, 73, 4806–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosgood, S.A.; Hoff, M.; Nicholson, M.L. Treatment of Transplant Kidneys during Machine Perfusion. Transpl. Int. Off. J. Eur. Soc. Organ. Transplant. 2021, 34, 224–232. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, M.R.; Wills, M.R.; Sinclair, J.H. HCMV Antivirals and Strategies to Target the Latent Reservoir. Viruses 2021, 13, 817. https://doi.org/10.3390/v13050817
Perera MR, Wills MR, Sinclair JH. HCMV Antivirals and Strategies to Target the Latent Reservoir. Viruses. 2021; 13(5):817. https://doi.org/10.3390/v13050817
Chicago/Turabian StylePerera, Marianne R., Mark R. Wills, and John H. Sinclair. 2021. "HCMV Antivirals and Strategies to Target the Latent Reservoir" Viruses 13, no. 5: 817. https://doi.org/10.3390/v13050817
APA StylePerera, M. R., Wills, M. R., & Sinclair, J. H. (2021). HCMV Antivirals and Strategies to Target the Latent Reservoir. Viruses, 13(5), 817. https://doi.org/10.3390/v13050817