
A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Isolation
2.2. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.3. Quantitative Real-Time RT-PCR (RT-qPCR)
2.4. Rapid Amplification of cDNA-Ends (RACE) with PCR
2.5. Phylogenetic and Sequence Analysis
3. Results
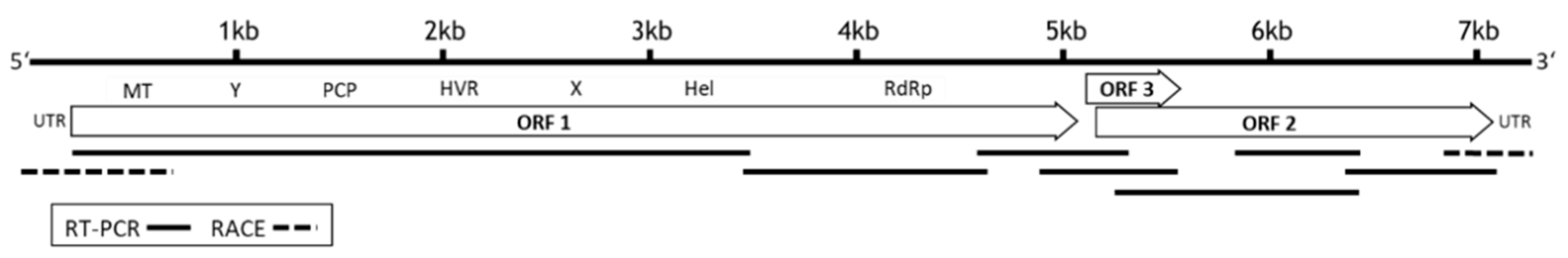
3.1. Full Genome Sequencing and Determination of Genome Organization
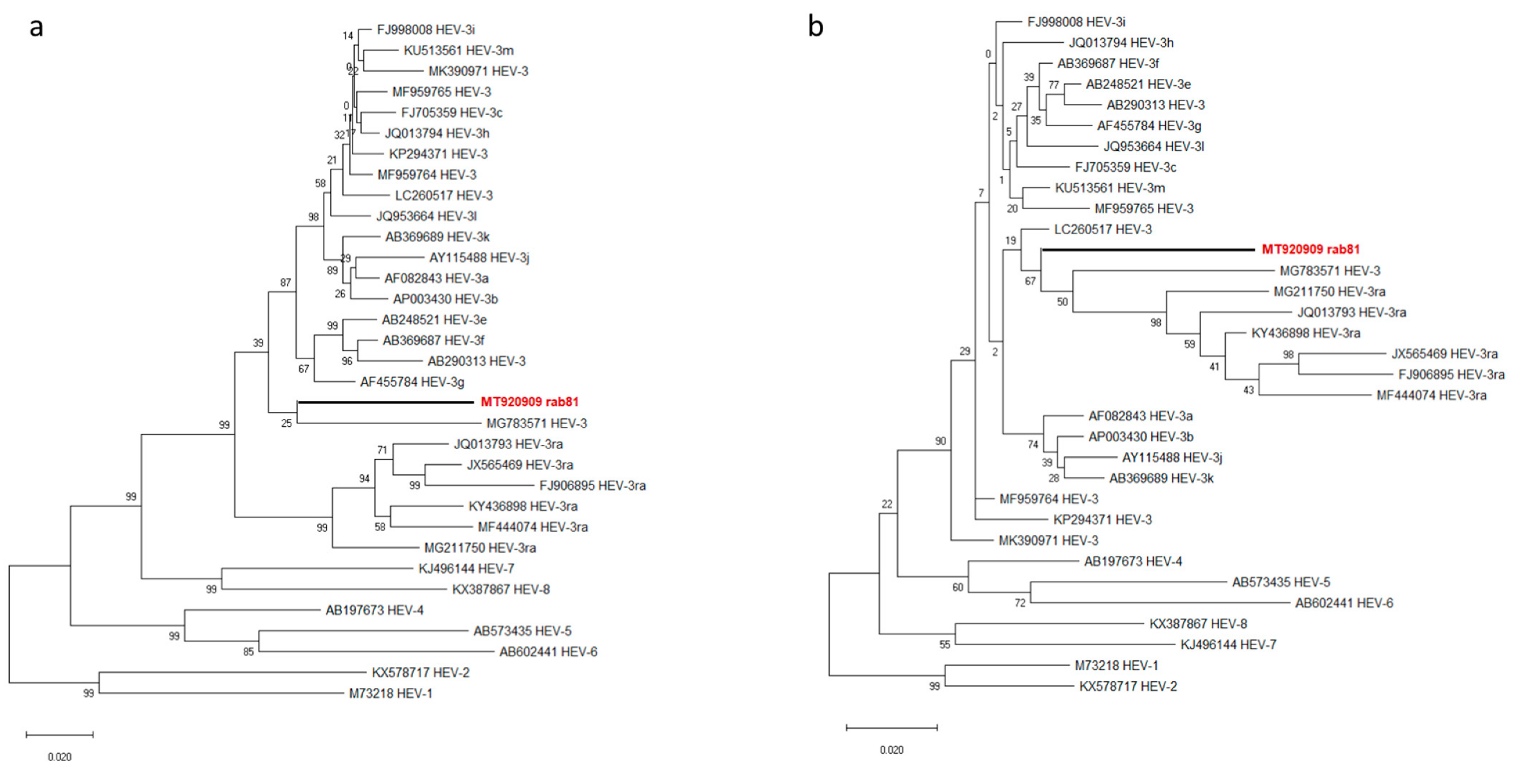
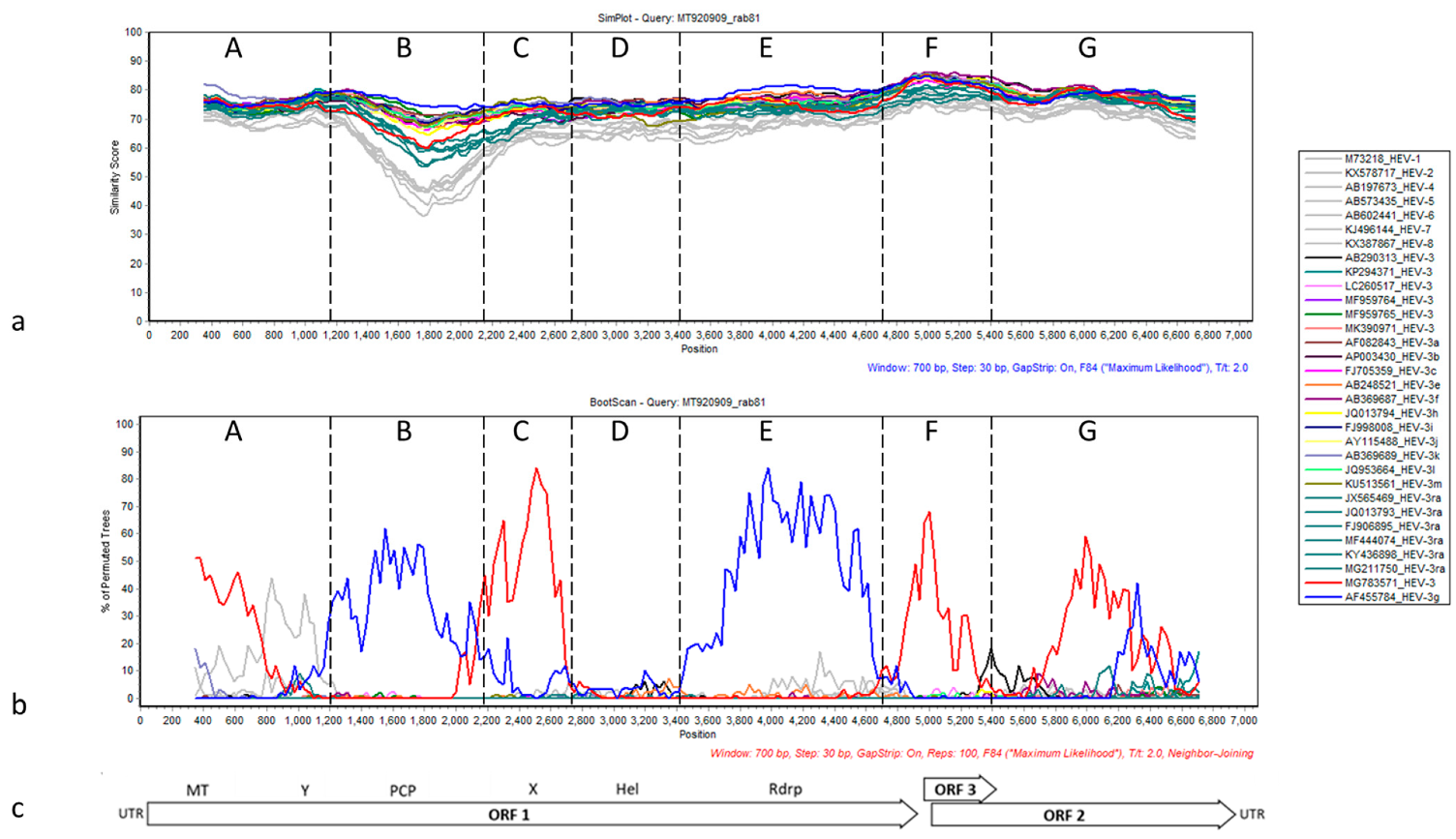
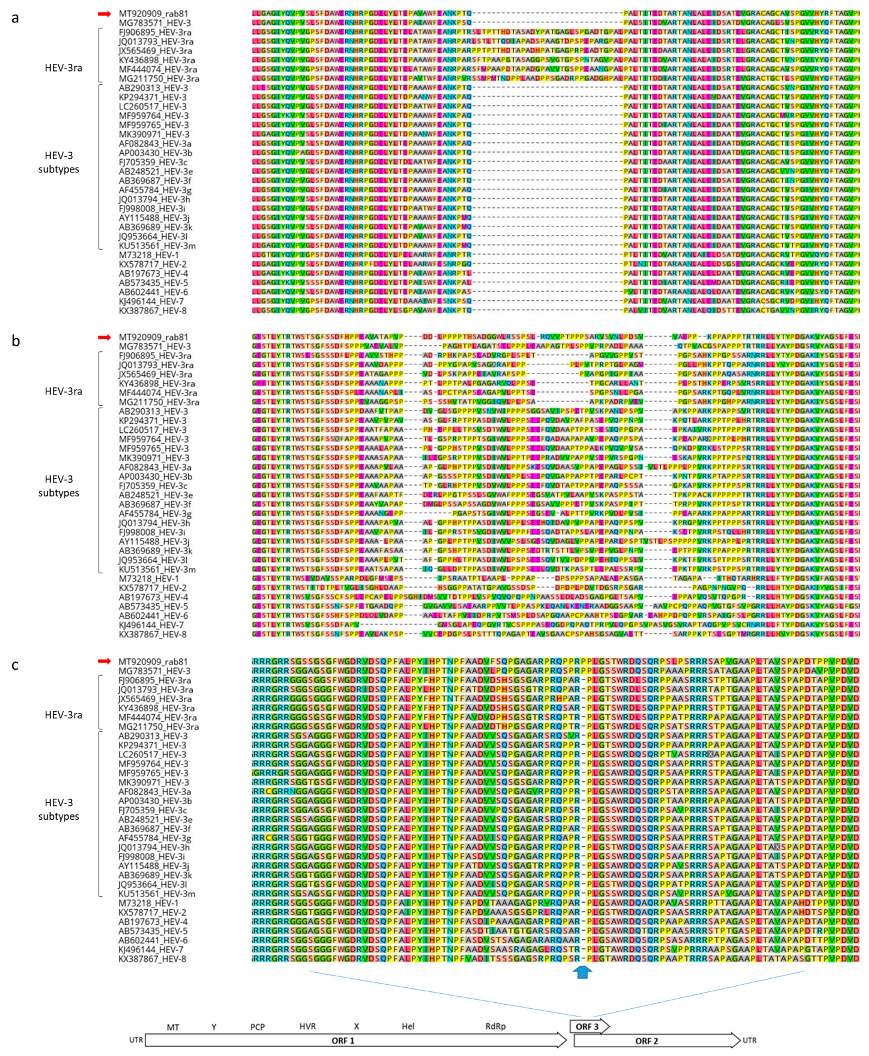
3.2. Sequence and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nelson, K.E.; Heaney, C.D.; Labrique, A.B.; Kmush, B.L.; Krain, L.J. Hepatitis E: Prevention and treatment. Curr. Opin. Infect. Dis. 2016, 29, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724. [Google Scholar] [CrossRef]
- Himmelsbach, K.; Bender, D.; Hildt, E. Life cycle and morphogenesis of the hepatitis E virus. Emerg. Microbes Infect. 2016, 7, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhomme, S.; Marion, O.; Abravanel, F.; Chapuy-Regaud, S.; Kamar, N.; Izopet, J. Hepatitis E Pathogenesis. Viruses 2016, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Rein, D.B.; Stevens, G.A.; Theaker, J.; Wittenborn, J.S.; Wiersma, S.T. The global burden of hepatitis E virus genotypes 1 and 2 in 2005. Hepatology 2012, 55, 988–997. [Google Scholar] [CrossRef]
- Spahr, C.; Knauf-Witzens, T.; Vahlenkamp, T.; Ulrich, R.G.; Johne, R. Hepatitis E virus and related viruses in wild, domestic and zoo animals: A review. Zoonoses Public Health 2018, 65, 11–29. [Google Scholar] [CrossRef]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef]
- Emerson, S.U.; Zhang, M.; Meng, X.J.; Nguyen, H.; St Claire, M.; Govindarajan, S.; Huang, Y.K.; Purcell, R.H. Recombinant hepatitis E virus genomes infectious for primates: Importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. USA 2001, 98, 15270–15275. [Google Scholar] [CrossRef] [Green Version]
- Dalton, H.R.; Izopet, J. Transmission and Epidemiology of Hepatitis E Virus Genotype 3 and 4 Infections. Cold Spring Harb. Perspect. Med. 2018, 8, a032144. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Zhao, Q.; Zhou, E.M. Zoonotic Hepatitis E Virus: An Ignored Risk for Public Health. Front. Microbiol. 2017, 8, 2396. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Ma, Z.; Harrison, T.J.; Feng, R.; Zhang, C.; Qiao, Z.; Fan, J.; Ma, H.; Li, M.; Song, A.; et al. A novel genotype of hepatitis E virus prevalent among farmed rabbits in China. J. Med. Virol. 2009, 81, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, F.; Schwaiger, K.; Dahnert, L.; Vina-Rodriguez, A.; Hoper, D.; Gareis, M.; Groschup, M.H.; Eiden, M. Hepatitis E virus in wild rabbits and European brown hares in Germany. Zoonoses Public Health 2017, 64, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Burt, S.A.; Veltman, J.; Hakze-van der Honing, R.; Schmitt, H.; van der Poel, W.H. Hepatitis E Virus in Farmed Rabbits, Wild Rabbits and Petting Farm Rabbits in the Netherlands. Food Environ. Virol. 2016, 8, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guérin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Cossaboom, C.M.; Cordoba, L.; Dryman, B.A.; Meng, X.J. Hepatitis E virus in rabbits, Virginia, USA. Emerg. Infect. Dis. 2011, 17, 2047–2049. [Google Scholar] [CrossRef] [PubMed]
- Sahli, R.; Fraga, M.; Semela, D.; Moradpour, D.; Gouttenoire, J. Rabbit HEV in immunosuppressed patients with hepatitis E acquired in Switzerland. J. Hepatol. 2019, 70, 1023–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulf, M.G.; Maguire, S.; Humbert, P.; Dai, N.; Bei, Y.; Nichols, N.M.; Corrêa, I.R.J.; Guan, S. Non-templated addition and template switching by Moloney murine leukemia virus (MMLV)-based reverse transcriptases co-occur and compete with each other. J. Biol. Chem. 2019, 294, 18220–18231. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vina-Rodriguez, A.; Schlosser, J.; Becher, D.; Kaden, V.; Groschup, M.H.; Eiden, M. Hepatitis E virus genotype 3 diversity: Phylogenetic analysis and presence of subtype 3b in wild boar in Europe. Viruses 2015, 7, 2704–2726. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Coller, K.E.; Dawson, G.J.; Cloherty, G.A. Identification of a putative novel genotype 3/rabbit hepatitis E virus (HEV) recombinant. PLoS ONE 2018, 13, e0203618. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.; Lopez-Lopez, P.; Zafra, I.; Caballero-Gomez, J.; Machuca, I.; Camacho, A.; Risalde, M.A.; Rivero-Juarez, A.; Rivero, A. Development and Clinical Validation of a Pangenotypic PCR-Based Assay for the Detection and Quantification of Hepatitis E Virus (Orthohepevirus A Genus). J. Clin. Microbiol. 2021, 59, e02075-20. [Google Scholar] [CrossRef]
- Cossaboom, C.M.; Córdoba, L.; Sanford, B.J.; Piñeyro, P.; Kenney, S.P.; Dryman, B.A.; Wang, Y.; Meng, X.J. Cross-species infection of pigs with a novel rabbit, but not rat, strain of hepatitis E virus isolated in the United States. J. Gen. Virol. 2012, 93, 1687–1695. [Google Scholar] [CrossRef]
- Liu, P.; Bu, Q.N.; Wang, L.; Han, J.; Du, R.J.; Lei, Y.X.; Ouyang, Y.Q.; Li, J.; Zhu, Y.H.; Lu, F.M.; et al. Transmission of hepatitis E virus from rabbits to cynomolgus macaques. Emerg. Infect. Dis. 2013, 19, 559–565. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; El Costa, H.; Schvartz, B.; Peron, J.M.; Kamar, N.; Izopet, J. Rabbit Hepatitis E Virus Infections in Humans, France. Emerg. Infect. Dis. 2017, 23, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Ryll, R.; Eiden, M.; Heuser, E.; Weinhardt, M.; Ziege, M.; Höper, D.; Groschup, M.H.; Heckel, G.; Johne, R.; Ulrich, R.G. Hepatitis E virus in feral rabbits along a rural-urban transect in Central Germany. Infect. Genet. Evol. 2018, 61, 155–159. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HEV-3 Subtype | Accession Number | Complete Genome | ORF1 | ORF2 | ORF3 | |||
|---|---|---|---|---|---|---|---|---|
| nt | nt | aa | nt | aa | nt | aa | ||
| HEV-3a | AF082843 | 78.6 | 77.4 | 89.8 | 81.6 | 93.0 | 90.4 | 87.7 |
| HEV-3b | AP003430 | 78.6 | 77.5 | 90.3 | 81.6 | 92.9 | 90.6 | 89.5 |
| HEV-3c | FJ705359 | 78.7 | 77.6 | 90.0 | 81.6 | 93.2 | 88.9 | 84.2 |
| HEV-3e | AB248521 | 79.3 | 78.1 | 90.2 | 82.1 | 93.5 | 88.9 | 86.8 |
| HEV-3f | AB369687 | 79.8 | 78.2 | 90.2 | 82.3 | 93.5 | 90.1 | 89.5 |
| HEV-3g | AF455784 | 80.0 | 79.2 | 90.7 | 82.3 | 92.9 | 89.2 | 88.6 |
| HEV-3h | JQ013794 | 78.6 | 77.3 | 90.4 | 81.9 | 93.2 | 91.5 | 89.5 |
| HEV-3i | FJ998008 | 79.0 | 77.9 | 89.8 | 81.9 | 93.6 | 90.4 | 90.4 |
| HEV-3j | AY115488 | 78.8 | 77.4 | 89.2 | 82.1 | 92.9 | 90.1 | 88.6 |
| HEV-3k | AB369689 | 79.8 | 78.9 | 90.7 | 81.9 | 92.9 | 90.7 | 90.4 |
| HEV-3l | JQ953664 | 79.2 | 77.9 | 90.2 | 82.0 | 93.2 | 90.4 | 90.4 |
| HEV-3m | KU513561 | 78.8 | 77.3 | 89.4 | 82.1 | 94.3 | 91.2 | 88.6 |
| HEV-3ra | FJ906895 | 75.3 | 73.4 | 84.8 | 79.8 | 89.7 | 83.8 | 80.7 |
| HEV-3 | AB290313 | 79.4 | 78.1 | 89.3 | 82.6 | 92.7 | 88.1 | 86.8 |
| HEV-3 | KP294371 | 79.4 | 78.2 | 89.6 | 82.4 | 92.4 | 87.8 | 86.8 |
| HEV-3 | LC260517 | 79.0 | 77.6 | 89.9 | 82.2 | 94.0 | 89.7 | 83.4 |
| HEV-3 | MF959764 | 79.1 | 78.1 | 89.9 | 81.7 | 93.5 | 89.3 | 86.0 |
| HEV-3 | MF959765 | 79.2 | 78.0 | 89.8 | 82.2 | 92.7 | 89.6 | 87.7 |
| HEV-3 | MK390971 | 79.1 | 77.7 | 89.8 | 82.6 | 92.6 | 89.3 | 86.8 |
| HEV-3 | MG783571 | 77.8 | 76.2 | 87.8 | 81.2 | 91.8 | 91.2 | 88.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cierniak, F.; von Arnim, F.; Heckel, G.; Ulrich, R.G.; Groschup, M.H.; Eiden, M. A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016. Viruses 2021, 13, 1065. https://doi.org/10.3390/v13061065
Cierniak F, von Arnim F, Heckel G, Ulrich RG, Groschup MH, Eiden M. A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016. Viruses. 2021; 13(6):1065. https://doi.org/10.3390/v13061065
Chicago/Turabian StyleCierniak, Filip, Felicitas von Arnim, Gerald Heckel, Rainer G. Ulrich, Martin H. Groschup, and Martin Eiden. 2021. "A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016" Viruses 13, no. 6: 1065. https://doi.org/10.3390/v13061065
APA StyleCierniak, F., von Arnim, F., Heckel, G., Ulrich, R. G., Groschup, M. H., & Eiden, M. (2021). A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016. Viruses, 13(6), 1065. https://doi.org/10.3390/v13061065