
Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses



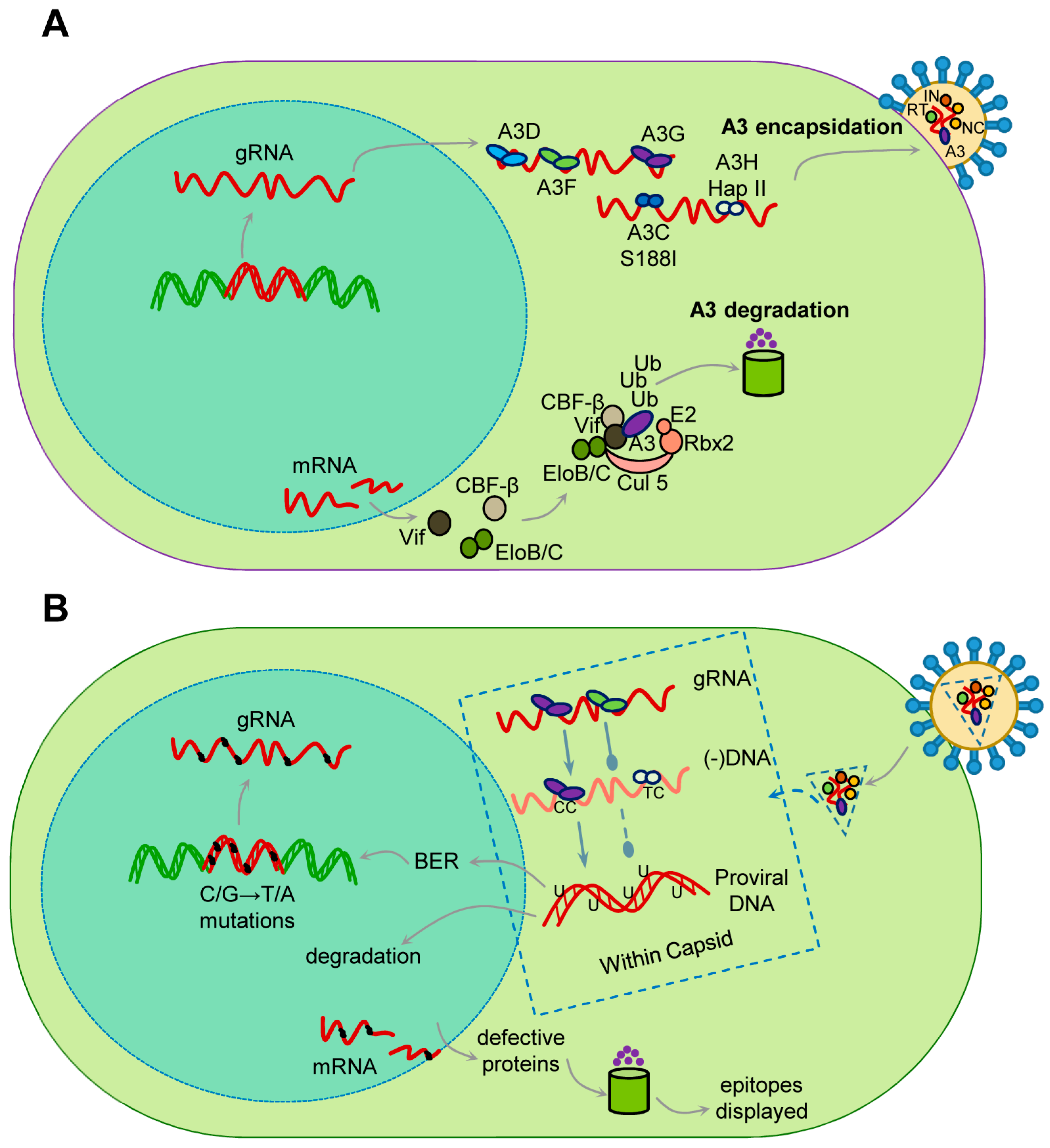{kind=link}
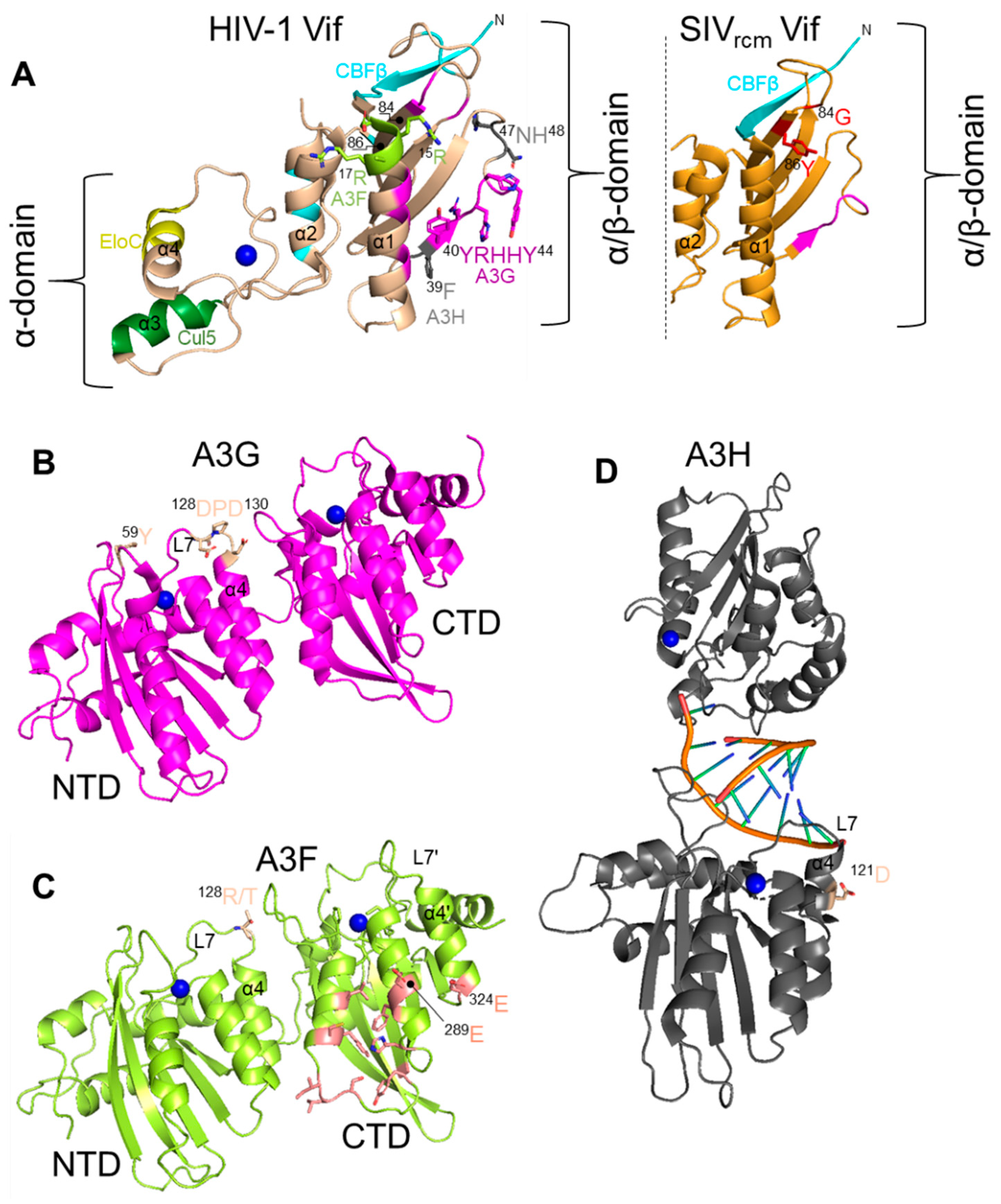{kind=link}
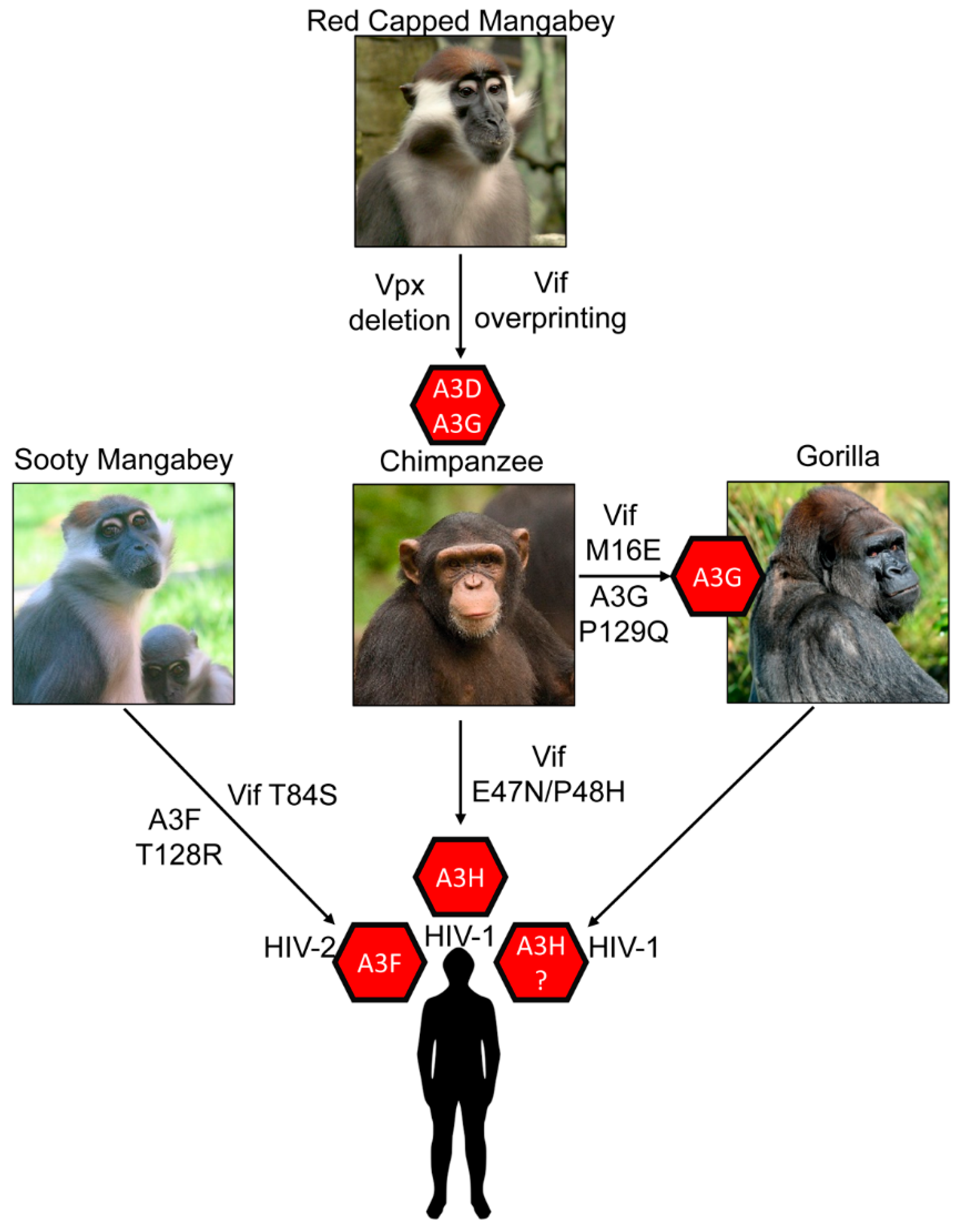{kind=link}
Abstract
:1. Introduction
2. Origin of the Immunodeficiency Virus in Humans
3. Lentiviral Restriction Factors and Their Viral Antagonists
4. Overview of APOBEC3s
5. The Vif-A3 Interaction and Cross Species Transmission
6. APOBEC3G
7. APOBEC3F
8. APOBEC3H
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [Green Version]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Borremans, B.; Faust, C.; Manlove, K.R.; Sokolow, S.H.; Lloyd-Smith, J.O. Cross-species pathogen spillover across ecosystem boundaries: Mechanisms and theory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180344. [Google Scholar] [CrossRef] [Green Version]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza a virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggal, N.K.; Emerman, M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. Immunol. 2012, 12, 687–695. [Google Scholar] [CrossRef]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell. Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011, 11, 45–56. [Google Scholar] [CrossRef]
- Sharp, P.M.; Bailes, E.; Chaudhuri, R.R.; Rodenburg, C.M.; Santiago, M.O.; Hahn, B.H. The origins of acquired immune deficiency syndrome viruses: Where and when? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 867–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, A.; Rawls, D.; Moore, J. Origin of HIV type 1 in colonial French Equatorial Africa? AIDS Res. Hum. Retrovir. 2000, 16, 5–8. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. The evolution of HIV-1 and the origin of AIDS. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2010, 365, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Bedford, T. Modern-day SIV viral diversity generated by extensive recombination and cross-species transmission. PLoS Pathog. 2017, 13, e1006466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayouba, A.; Akoua-Koffi, C.; Calvignac-Spencer, S.; Esteban, A.; Locatelli, S.; Li, H.; Li, Y.; Hahn, B.H.; Delaporte, E.; Leendertz, F.H.; et al. Evidence for continuing cross-species transmission of SIVsmm to humans: Characterization of a new HIV-2 lineage in rural Cote d’Ivoire. AIDS 2013, 27, 2488–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neil, S.J.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Sauter, D.; Kirchhoff, F. Tetherin antagonism by primate lentiviral nef proteins. Curr. HIV Res. 2011, 9, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Nakano, Y.; Aso, H.; Soper, A.; Yamada, E.; Moriwaki, M.; Juarez-Fernandez, G.; Koyanagi, Y.; Sato, K. A conflict of interest: The evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology 2017, 14, 31. [Google Scholar] [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; White, T.E.; Schulte, B.; de Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nunez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Thomas, C.J.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC family of polynucleotide deoxy)cytidine deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Baig, T.T.; Love, R.P.; Chelico, L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 2014, 5, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, M.B.; Love, R.P.; Chelico, L. Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infect. Dis. 2018, 4, 224–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E.; et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 2019, 4, 78–88. [Google Scholar] [CrossRef]
- Arias, J.F.; Koyama, T.; Kinomoto, M.; Tokunaga, K. Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbiol. 2012, 3, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqassim, E.Y.; Sharma, S.; Khan, A.; Emmons, T.R.; Cortes Gomez, E.; Alahmari, A.; Singel, K.L.; Mark, J.; Davidson, B.A.; Robert McGray, A.J.; et al. RNA editing enzyme APOBEC3A promotes pro-inflammatory M1 macrophage polarization. Commun. Biol. 2021, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Kosugi, Y.; Ito, J.; Sato, K. The Battle between Retroviruses and APOBEC3 Genes: Its Past and Present. Viruses 2021, 13, 124. [Google Scholar] [CrossRef]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.S. Insights into the Structures and Multimeric Status of APOBEC Proteins Involved in Viral Restriction and Other Cellular Functions. Viruses 2021, 13, 497. [Google Scholar] [CrossRef]
- LaRue, R.S.; Jonsson, S.R.; Silverstein, K.A.; Lajoie, M.; Bertrand, D.; El-Mabrouk, N.; Hotzel, I.; Andresdottir, V.; Smith, T.P.; Harris, R.S. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huthoff, H.; Malim, M.H. Cytidine deamination and resistance to retroviral infection: Towards a structural understanding of the APOBEC proteins. Virology 2005, 334, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, M.; Brayton, B.; Letko, M.; Maio, S.M.; Pilcher, C.D.; Hecht, F.M.; Barbour, J.D.; Simon, V. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 2013, 14, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Y.; Wang, X.; Esselman, W.J.; Zheng, Y.H. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 2006, 80, 10522–10533. [Google Scholar] [CrossRef] [Green Version]
- OhAinle, M.; Kerns, J.A.; Li, M.M.; Malik, H.S.; Emerman, M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008, 4, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refsland, E.W.; Hultquist, J.F.; Luengas, E.M.; Ikeda, T.; Shaban, N.M.; Law, E.K.; Brown, W.L.; Reilly, C.; Emerman, M.; Harris, R.S. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet. 2014, 10, e1004761. [Google Scholar] [CrossRef]
- Wittkopp, C.J.; Adolph, M.B.; Wu, L.I.; Chelico, L.; Emerman, M. A Single Nucleotide Polymorphism in Human APOBEC3C Enhances Restriction of Lentiviruses. PLoS Pathog. 2016, 12, e1005865. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Emerman, M. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J. Virol. 2011, 85, 8197–8207. [Google Scholar] [CrossRef] [Green Version]
- Chesarino, N.M.; Emerman, M. Polymorphisms in Human APOBEC3H Differentially Regulate Ubiquitination and Antiviral Activity. Viruses 2020, 12, 378. [Google Scholar] [CrossRef] [Green Version]
- Harari, A.; Ooms, M.; Mulder, L.C.; Simon, V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 2009, 83, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, M.; Majdak, S.; Seibert, C.W.; Harari, A.; Simon, V. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J. Virol. 2010, 84, 7961–7969. [Google Scholar] [CrossRef] [Green Version]
- Adolph, M.B.; Ara, A.; Feng, Y.; Wittkopp, C.J.; Emerman, M.; Fraser, J.S.; Chelico, L. Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res. 2017, 45, 3378–3394. [Google Scholar] [CrossRef]
- Apolonia, L.; Schulz, R.; Curk, T.; Rocha, P.; Swanson, C.M.; Schaller, T.; Ule, J.; Malim, M.H. Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PLoS Pathog. 2015, 11, e1004609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- York, A.; Kutluay, S.B.; Errando, M.; Bieniasz, P.D. The RNA Binding Specificity of Human APOBEC3 Proteins Resembles That of HIV-1 Nucleocapsid. PLoS Pathog. 2016, 12, e1005833. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, W.; Tian, C.; Liu, B.; Yu, Y.; Ding, L.; Spearman, P.; Yu, X.F. Distinct viral determinants for the packaging of human cytidine deaminases APOBEC3G and APOBEC3C. Virology 2008, 377, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Greenwell-Wild, T.; Nares, S.; Jin, W.; Lei, K.J.; Rangel, Z.G.; Munson, P.J.; Wahl, S.M. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 2007, 110, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguiar, R.S.; Lovsin, N.; Tanuri, A.; Peterlin, B.M. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2008, 283, 2518–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, V.; Heidecker, G.; Pathak, V.K.; Derse, D. The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J. Virol. 2011, 85, 8538–8547. [Google Scholar] [CrossRef] [Green Version]
- Salamango, D.J.; McCann, J.L.; Demir, O.; Brown, W.L.; Amaro, R.E.; Harris, R.S. APOBEC3B Nuclear Localization Requires Two Distinct N-Terminal Domain Surfaces. J. Mol. Biol. 2018, 430, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Stewart-Maynard, K.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [Green Version]
- Adolph, M.B.; Webb, J.; Chelico, L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PLoS ONE 2013, 8, e64196. [Google Scholar]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Li, X.; Wang, D.; Cui, Z.; Li, Q.; Li, M.; Ma, Y.; Hu, Q.; Zhou, Y.; Zhang, X.E. HIV-1 viral cores enter the nucleus collectively through the nuclear endocytosis-like pathway. Sci. China Life Sci. 2021, 64, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep. 2020, 32, 108201. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, N.; Love, R.P.; Gibson, R.; Arts, E.J.; Poon, A.F.Y.; Chelico, L. Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance. Heliyon 2019, 5, e01498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506.e494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef]
- Bogerd, H.P.; Tallmadge, R.L.; Oaks, J.L.; Carpenter, S.; Cullen, B.R. Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J. Virol. 2008, 82, 11889–11901. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Bergeron, J.R.; Huthoff, H.; Veselkov, D.A.; Beavil, R.L.; Simpson, P.J.; Matthews, S.J.; Malim, M.H.; Sanderson, M.R. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 2010, 6, e1000925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Xiao, Z.; Ehrlich, E.; Yu, Y.; Liu, B.; Zheng, S.; Yu, X.F. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 2005, 102, 11444–11449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangeat, B.; Turelli, P.; Liao, S.; Trono, D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J. Biol. Chem. 2004, 279, 14481–14483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, B.J.; Ehrlich, E.S.; Short, L.; Yu, Y.; Xiao, Z.; Yu, X.F.; Xiong, Y. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J. Virol. 2008, 82, 8656–8663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Binka, M.; LaRue, R.S.; Simon, V.; Harris, R.S. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J. Virol. 2012, 86, 2874–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Evans, S.L.; Han, X.; Liu, Y.; Yu, X.F. Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFbeta and CRL5 E3 ubiquitin ligase components. PLoS ONE 2012, 7, e33495. [Google Scholar]
- Zhang, W.; Wang, H.; Li, Z.; Liu, X.; Liu, G.; Harris, R.S.; Yu, X.F. Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J. Virol. 2014, 88, 12528–12540. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, R.; Izumi, T.; Nakano, Y.; Yamada, E.; Moriwaki, M.; Misawa, N.; Ren, F.; Kobayashi, T.; Koyanagi, Y.; Sato, K. Small ruminant lentiviral Vif proteins commonly utilize cyclophilin A, an evolutionarily and structurally conserved protein, to degrade ovine and caprine APOBEC3 proteins. Microbiol. Immunol. 2016, 60, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Zhu, D.; Wang, C.; Su, C.; Ma, J.; Ma, J.; Wang, X. Core-binding factor subunit beta is not required for non-primate lentiviral Vif-mediated APOBEC3 degradation. J. Virol. 2014, 88, 12112–12122. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 2013, 49, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, M.; Zhang, F.; Bieniasz, P.D. Single-Cell and Single-Cycle Analysis of HIV-1 Replication. PLoS Pathog. 2015, 11, e1004961. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef] [Green Version]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 5501–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Etienne, L.; Hahn, B.H.; Sharp, P.M.; Matsen, F.A.; Emerman, M. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 2013, 14, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Gu, Q.; de Manuel Montero, M.; Bravo, I.G.; Marques-Bonet, T.; Haussinger, D.; Munk, C. Stably expressed APOBEC3H forms a barrier for cross-species transmission of simian immunodeficiency virus of chimpanzee to humans. PLoS Pathog. 2017, 13, e1006746. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Kirchhoff, F. Key Viral Adaptations Preceding the AIDS Pandemic. Cell Host Microbe 2019, 25, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Munch, J.; Kim, K.A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 2009, 6, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Plantier, J.C.; Leoz, M.; Dickerson, J.E.; De Oliveira, F.; Cordonnier, F.; Lemee, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delviks-Frankenberry, K.A.; Desimmie, B.A.; Pathak, V.K. Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses 2020, 12, 587. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Knecht, K.M.; Shen, Q.; Xiong, Y. Multifaceted HIV-1 Vif interactions with human E3 ubiquitin ligase and APOBEC3s. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Schröfelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef] [Green Version]
- Bogerd, H.P.; Doehle, B.P.; Wiegand, H.L.; Cullen, B.R. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. USA 2004, 101, 3770–3774. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Svarovskaia, E.S.; Barr, R.; Zhang, Y.; Khan, M.A.; Strebel, K.; Pathak, V.K. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. USA 2004, 101, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Huthoff, H.; Malim, M.H. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J. Virol. 2007, 81, 3807–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavens, D.; Peelman, F.; Van der Heyden, J.; Uyttendaele, I.; Catteeuw, D.; Verhee, A.; Van Schoubroeck, B.; Kurth, J.; Hallenberger, S.; Clayton, R.; et al. Definition of the interacting interfaces of Apobec3G and HIV-1 Vif using MAPPIT mutagenesis analysis. Nucleic Acids Res. 2010, 38, 1902–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Love, R.P.; Chelico, L. HIV-1 viral infectivity factor (Vif) alters processive single-stranded DNA scanning of the retroviral restriction factor APOBEC3G. J. Biol. Chem. 2013, 288, 6083–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Silvestri, G.; Hahn, B.H.; Bibollet-Ruche, F.; Gokcumen, O.; Simon, V.; Ooms, M. Vif proteins from diverse primate lentiviral lineages use the same binding site in APOBEC3G. J. Virol. 2013, 87, 11861–11871. [Google Scholar] [CrossRef] [Green Version]
- Binning, J.M.; Chesarino, N.M.; Emerman, M.; Gross, J.D. Structural Basis for a Species-Specific Determinant of an SIV Vif Protein toward Hominid APOBEC3G Antagonism. Cell Host Microbe 2019, 26, 739–747.e734. [Google Scholar] [CrossRef]
- Russell, R.A.; Pathak, V.K. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 2007, 81, 8201–8210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Booiman, T.; Kootstra, N.; Simon, V.; Ooms, M. Identification of the HIV-1 Vif and Human APOBEC3G Protein Interface. Cell Rep. 2015, 13, 1789–1799. [Google Scholar] [CrossRef] [Green Version]
- Schrofelbauer, B.; Senger, T.; Manning, G.; Landau, N.R. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J. Virol. 2006, 80, 5984–5991. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.; Albin, J.S.; Demir, O.; Shaban, N.M.; Luengas, E.M.; Land, A.M.; Anderson, B.D.; Holten, J.R.; Anderson, J.S.; Harki, D.A.; et al. The Binding Interface between Human APOBEC3F and HIV-1 Vif Elucidated by Genetic and Computational Approaches. Cell Rep. 2015, 13, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S.; Anderson, B.D. Evolutionary Paradigms from Ancient and Ongoing Conflicts between the Lentiviral Vif Protein and Mammalian APOBEC3 Enzymes. PLoS Pathog. 2016, 12, e1005958. [Google Scholar] [CrossRef] [PubMed]
- Sobieszczyk, M.E.; Lingappa, J.R.; McElrath, M.J. Host genetic polymorphisms associated with innate immune factors and HIV-1. Curr Opin. HIV AIDS 2011, 6, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Emerman, M.; Malik, H.S. Paleovirology--modern consequences of ancient viruses. PLoS Biol. 2010, 8, e1000301. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef]
- Munk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012, 12, 71. [Google Scholar] [CrossRef] [Green Version]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, A.A.; Hirsch, V.M.; Emerman, M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe 2012, 11, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupp, A.; McCarthy, K.R.; Ooms, M.; Letko, M.; Morgan, J.S.; Simon, V.; Johnson, W.E. APOBEC3G polymorphism as a selective barrier to cross-species transmission and emergence of pathogenic SIV and AIDS in a primate host. PLoS Pathog. 2013, 9, e1003641. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Yamamoto, K.; Ueda, M.T.; Soper, A.; Konno, Y.; Kimura, I.; Uriu, K.; Kumata, R.; Aso, H.; Misawa, N.; et al. A role for gorilla APOBEC3G in shaping lentivirus evolution including transmission to humans. PLoS Pathog. 2020, 16, e1008812. [Google Scholar] [CrossRef]
- Chaipan, C.; Smith, J.L.; Hu, W.S.; Pathak, V.K. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 2013, 87, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Ara, A.; Love, R.P.; Chelico, L. Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 2014, 10, e1004024. [Google Scholar] [CrossRef] [Green Version]
- Mohammadzadeh, N.; Follack, T.B.; Love, R.P.; Stewart, K.; Sanche, S.; Chelico, L. Polymorphisms of the cytidine deaminase APOBEC3F have different HIV-1 restriction efficiencies. Virology 2019, 527, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Ara, A.; Love, R.P.; Follack, T.B.; Ahmed, K.A.; Adolph, M.B.; Chelico, L. Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albin, J.S.; Hache, G.; Hultquist, J.F.; Brown, W.L.; Harris, R.S. Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function. J. Virol. 2010, 84, 10209–10219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hache, G.; Shindo, K.; Albin, J.S.; Harris, R.S. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr Biol. 2008, 18, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Symeonides, M.; Albin, J.S.; Li, M.; Thali, M.; Harris, R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog. 2018, 14, e1007010. [Google Scholar] [CrossRef] [Green Version]
- Nchioua, R.; Kmiec, D.; Gaba, A.; Stürzel, C.M.; Follack, T.; Patrick, S.; Kirmaier, A.; Johnson, W.E.; Hahn, B.H.; Chelico, L.; et al. APOBEC3F constitutes a barrier to successful cross-species transmission of SIVsmm to humans. J. Virol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Smith, J.L.; Izumi, T.; Borbet, T.C.; Hagedorn, A.N.; Pathak, V.K. HIV-1 and HIV-2 Vif interact with human APOBEC3 proteins using completely different determinants. J. Virol. 2014, 88, 9893–9908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.A.; Smith, J.; Barr, R.; Bhattacharyya, D.; Pathak, V.K. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 2009, 83, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albin, J.S.; LaRue, R.S.; Weaver, J.A.; Brown, W.L.; Shindo, K.; Harjes, E.; Matsuo, H.; Harris, R.S. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J. Biol. Chem. 2010, 285, 40785–40792. [Google Scholar] [CrossRef] [Green Version]
- Zhen, A.; Wang, T.; Zhao, K.; Xiong, Y.; Yu, X.F. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 2010, 84, 1902–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Desimmie, B.A.; Nguyen, H.C.; Ziegler, S.J.; Cheng, T.C.; Chen, J.; Wang, J.; Wang, H.; Zhang, K.; Pathak, V.K.; et al. Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat. Struct. Mol. Biol. 2019, 26, 1176–1183. [Google Scholar] [CrossRef]
- Dang, Y.; Siew, L.M.; Wang, X.; Han, Y.; Lampen, R.; Zheng, Y.H. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 2008, 283, 11606–11614. [Google Scholar] [CrossRef] [Green Version]
- OhAinle, M.; Kerns, J.A.; Malik, H.S.; Emerman, M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 2006, 80, 3853–3862. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Abudu, A.; Son, S.; Dang, Y.; Venta, P.J.; Zheng, Y.H. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 2011, 85, 3142–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Wong, L.; Morse, M.; Rouzina, I.; Williams, M.C.; Chelico, L. RNA-Mediated Dimerization of the Human Deoxycytidine Deaminase APOBEC3H Influences Enzyme Activity and Interaction with Nucleic Acids. J. Mol. Biol. 2018, 430, 4891–4907. [Google Scholar] [CrossRef]
- Ito, F.; Yang, H.; Xiao, X.; Li, S.X.; Wolfe, A.; Zirkle, B.; Arutiunian, V.; Chen, X.S. Understanding the Structure, Multimerization, Subcellular Localization and mC Selectivity of a Genomic Mutator and Anti-HIV Factor APOBEC3H. Sci. Rep. 2018, 8, 3763. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Nagae, T.; Ode, H.; Awazu, H.; Kurosawa, T.; Hamano, A.; Matsuoka, K.; Hachiya, A.; Imahashi, M.; Yokomaku, Y.; et al. Structural basis of chimpanzee APOBEC3H dimerization stabilized by double-stranded RNA. Nucleic Acids Res. 2018, 46, 10368–10379. [Google Scholar] [CrossRef] [Green Version]
- Bohn, J.A.; Thummar, K.; York, A.; Raymond, A.; Brown, W.C.; Bieniasz, P.D.; Hatziioannou, T.; Smith, J.L. APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat. Commun. 2017, 8, 1021. [Google Scholar] [CrossRef] [Green Version]
- Shaban, N.M.; Shi, K.; Lauer, K.V.; Carpenter, M.A.; Richards, C.M.; Salamango, D.; Wang, J.; Lopresti, M.W.; Banerjee, S.; Levin-Klein, R.; et al. The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol. Cell 2018, 69, 75–86.e79. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Kurosawa, T.; Yokomaku, Y.; Yamane, T.; Watanabe, N.; Suzuki, A.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. [Google Scholar] [CrossRef]
- Baig, T.T.; Feng, Y.; Chelico, L. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. J. Virol. 2014, 88, 14380–14395. [Google Scholar] [CrossRef] [Green Version]
- Simon, V.; Zennou, V.; Murray, D.; Huang, Y.; Ho, D.D.; Bieniasz, P.D. Natural variation in Vif: Differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005, 1, e6. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, M.M.; Fahrny, A.; Jayaprakash, A.; Gers-Huber, G.; Dillon-White, M.; Audige, A.; Mulder, L.C.F.; Sachidanandam, R.; Speck, R.F.; Simon, V. Impact of Suboptimal APOBEC3G Neutralization on the Emergence of HIV Drug Resistance in Humanized Mice. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Monajemi, M.; Woodworth, C.F.; Zipperlen, K.; Gallant, M.; Grant, M.D.; Larijani, M. Positioning of APOBEC3G/F mutational hotspots in the human immunodeficiency virus genome favors reduced recognition by CD8+ T cells. PLoS ONE 2014, 9, e93428. [Google Scholar]
- Casartelli, N.; Guivel-Benhassine, F.; Bouziat, R.; Brandler, S.; Schwartz, O.; Moris, A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 2010, 207, 39–49. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.A.; Nikolaitchik, O.A.; Burdick, R.C.; Gorelick, R.J.; Keele, B.F.; Hu, W.S.; Pathak, V.K. Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS Pathog. 2016, 12, e1005646. [Google Scholar] [CrossRef] [Green Version]
- Lobritz, M.A.; Lassen, K.G.; Arts, E.J. HIV-1 replicative fitness in elite controllers. Curr. Opin. HIV AIDS 2011, 6, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Henry, K.R.; Weber, J.; Quinones-Mateu, M.E.; Arts, E.J. The impact of viral and host elements on HIV fitness and disease progression. Curr. HIV/AIDS Rep. 2007, 4, 36–41. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLoS Pathog. 2020, 16, e1008718. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Tachedjian, M.; Cui, J.; Cheng, A.Z.; Johnson, A.; Baker, M.L.; Harris, R.S.; Wang, L.F.; Tachedjian, G. Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol. Biol. Evol. 2018, 35, 1626–1637. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenbucher, A.; Bowen, D.; Sakhtemani, R.; Bournique, E.; Wise, J.F.; Zou, L.; Bhagwat, A.S.; Buisson, R.; Lawrence, M.S. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021, 12, 1602. [Google Scholar] [CrossRef]
- Starrett, G.J.; Luengas, E.M.; McCann, J.L.; Ebrahimi, D.; Temiz, N.A.; Love, R.P.; Feng, Y.; Adolph, M.B.; Chelico, L.; Law, E.K.; et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 2016, 7, 12918. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaba, A.; Flath, B.; Chelico, L. Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses. Viruses 2021, 13, 1084. https://doi.org/10.3390/v13061084
Gaba A, Flath B, Chelico L. Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses. Viruses. 2021; 13(6):1084. https://doi.org/10.3390/v13061084
Chicago/Turabian StyleGaba, Amit, Ben Flath, and Linda Chelico. 2021. "Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses" Viruses 13, no. 6: 1084. https://doi.org/10.3390/v13061084
APA StyleGaba, A., Flath, B., & Chelico, L. (2021). Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses. Viruses, 13(6), 1084. https://doi.org/10.3390/v13061084