
Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses
2.3. PDGFRα-Derived Soluble Receptor and Peptides
2.4. Selection of Viruses with Resistance against PDGFRα-Fc or the PDGFRα-Derived Peptides IK40 and GT40
2.5. Dose–Response Curves
2.6. Detection of Viral Immediate Early Proteins by Indirect Immunofluorescence
2.7. Generation of Mutant Viruses
2.8. Retransformation of E. coli with Viral Genomes and Analysis of the DNA Sequence
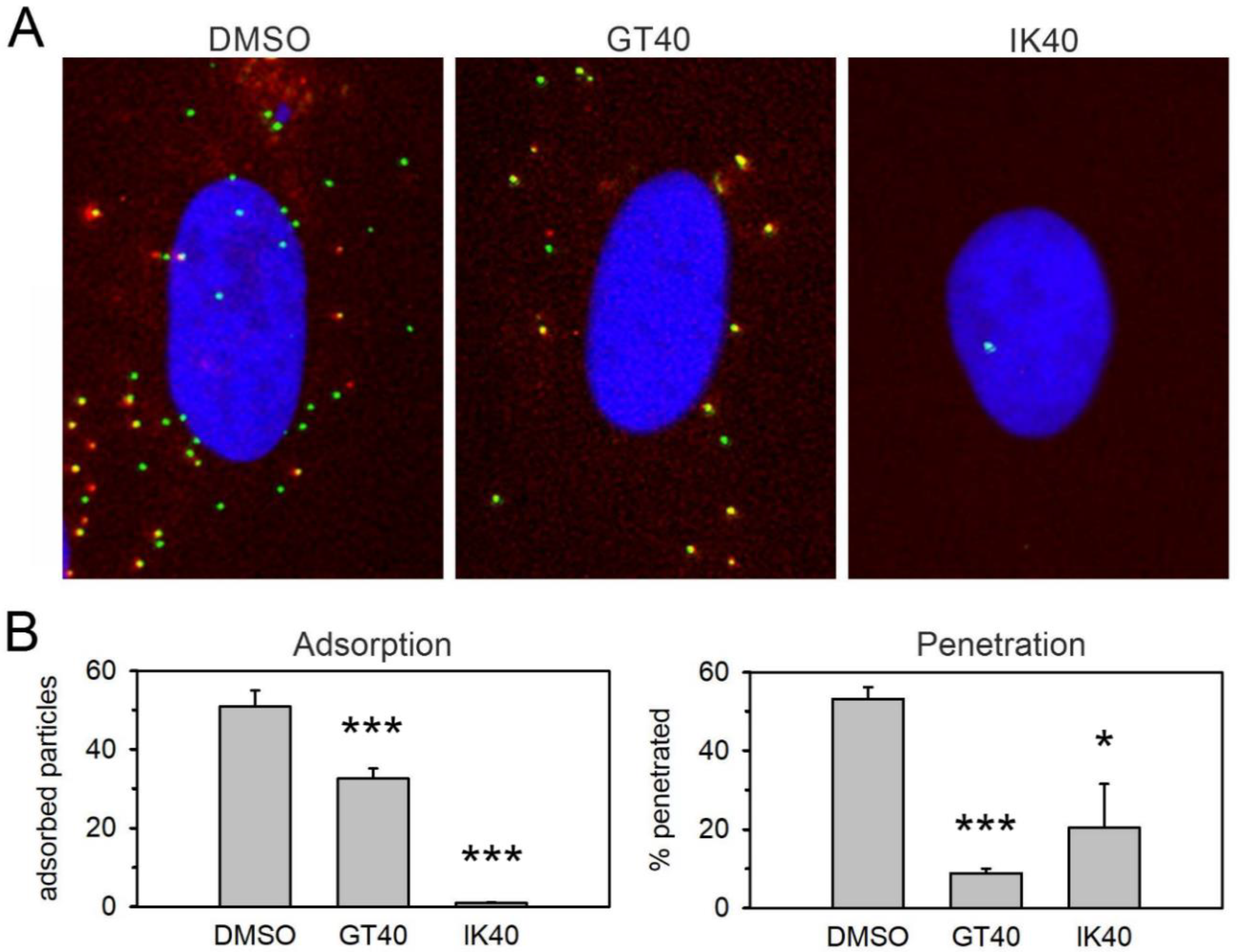
2.9. Quantification of Adsorption and Penetration
2.10. Statistical Analyses
3. Results
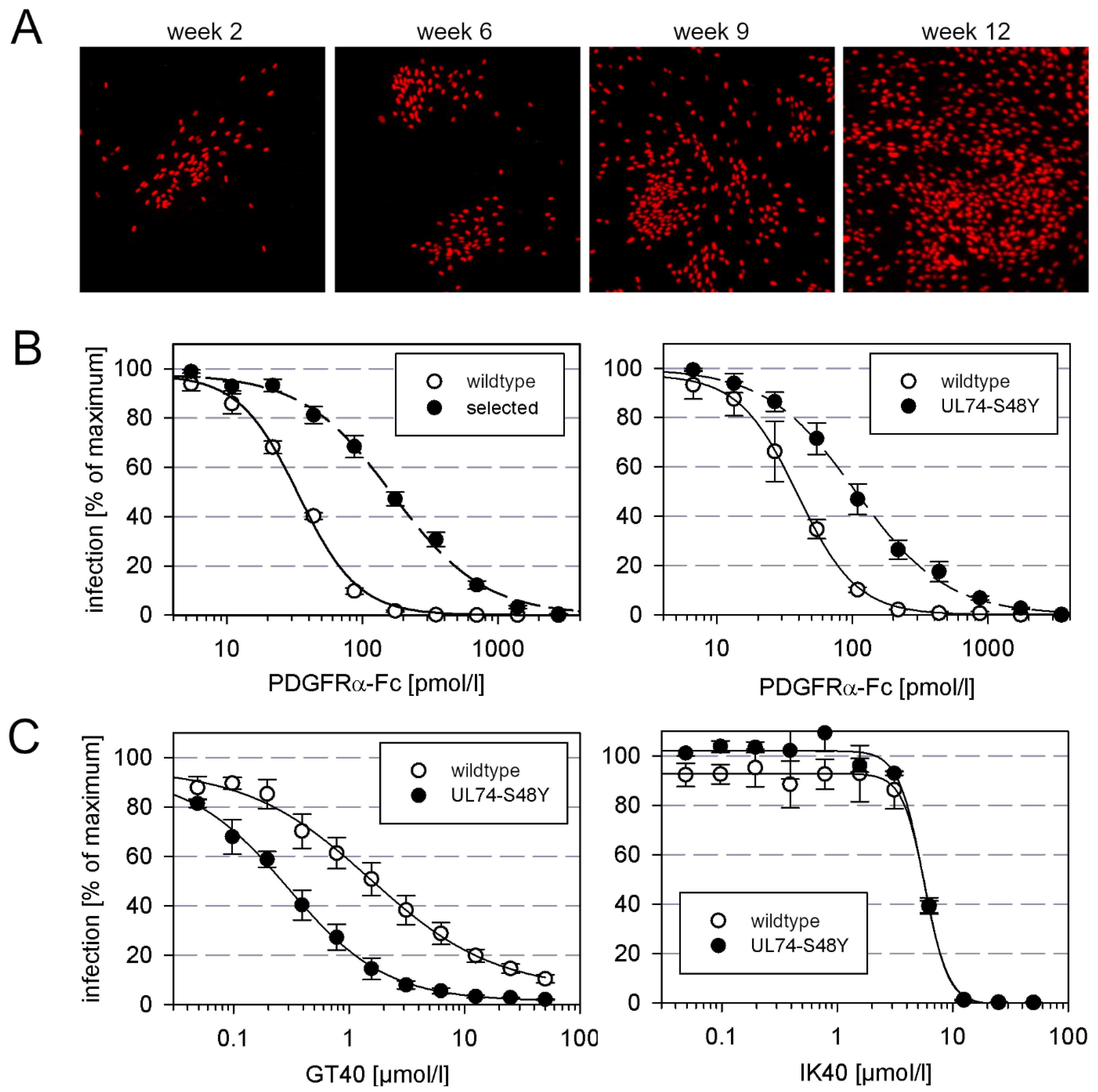
3.1. Selection of an AD169 Mutant with Decreased Sensitivity to the Entry Inhibitor PDGFRα-Fc
3.2. Marker Transfer Confirms That the S48Y Mutation in the N-Terminus of gO Confers Partial Resistance against PDGFRα-Fc
3.3. The PDGFRα-Derived Peptide GT40 Resembles Full-Length PDGFRα-Fc Regarding the Mode of Action
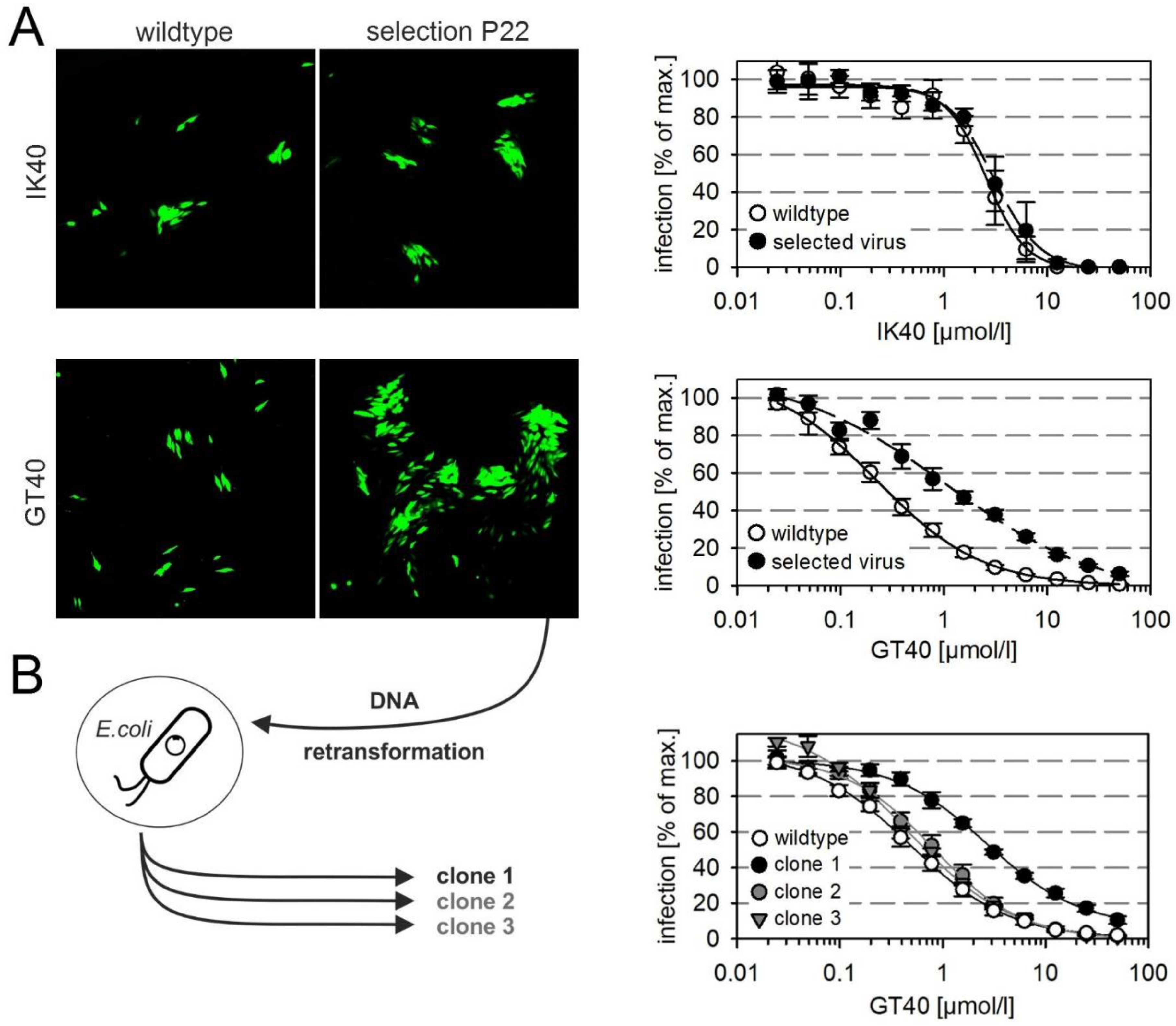
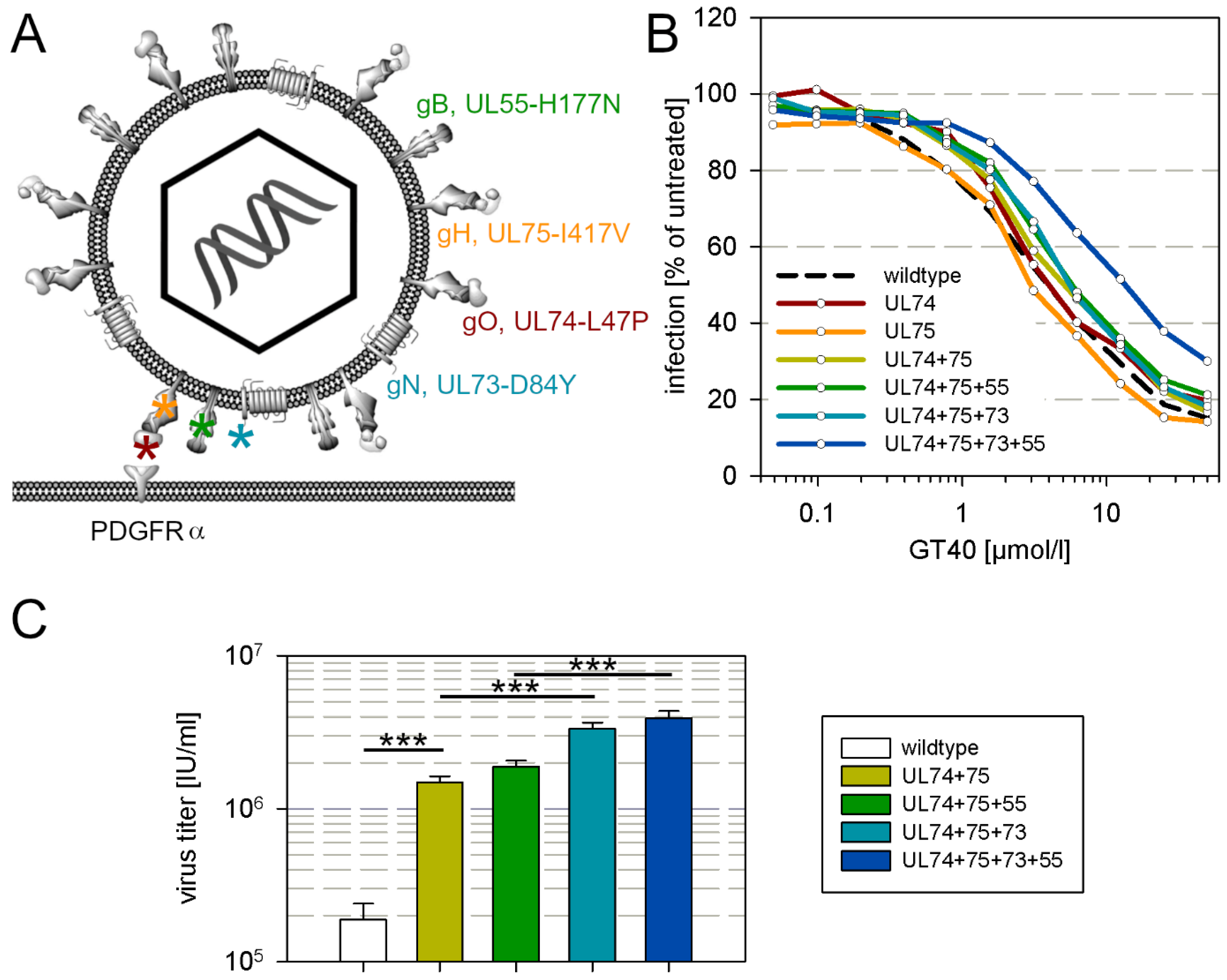
3.4. Selection of a Mutant with Decreased Sensitivity to the PDGFRα-Derived Peptide GT40
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Razonable, R.R. Drug-resistant cytomegalovirus: Clinical implications of specific mutations. Curr. Opin. Organ Transplant. 2018, 23, 388–394. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Lischka, P.; Michel, D.; Zimmermann, H. Characterization of Cytomegalovirus Breakthrough Events in a Phase 2 Prophylaxis Trial of Letermovir (AIC246, MK 8228). J. Infect. Dis. 2016, 213, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, R.M. Investigational Antiretroviral Drugs: What is Coming Down the Pipeline. Top. Antivir. Med. 2018, 25, 127–132. [Google Scholar]
- Haqqani, A.A.; Tilton, J.C. Entry inhibitors and their use in the treatment of HIV-1 infection. Antivir. Res. 2013, 98, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, C.; Hochdorfer, D.; Lieber, D.; Subramanian, N.; Stöhr, D.; Laib Sampaio, K.; Sinzger, C. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog. 2017, 13, e1006273. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, S.; Grimm, I.; Stöhr, D.; Antonini, C.; Lischka, P.; Sinzger, C.; Stegmann, C. Targeted mutagenesis on PDGFRα-Fc identifies amino acid modifications that allow efficient inhibition of HCMV infection while abolishing PDGF sequestration. PLoS Pathog. 2021, 17, e1009471. [Google Scholar] [CrossRef]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef] [Green Version]
- Borst, E.M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: A new approach for construction of HCMV mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. Clifton NJ 2010, 634, 421–430. [Google Scholar]
- Laib Sampaio, K.; Weyell, A.; Subramanian, N.; Wu, Z.; Sinzger, C. A TB40/E-derived human cytomegalovirus genome with an intact US-gene region and a self-excisable BAC cassette for immunological research. BioTechniques 2017, 63, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.J.; Sampaio, K.L.; Ettischer, N.; Stierhof, Y.-D.; Jahn, G.; Kropff, B.; Mach, M.; Sinzger, C. UL74 of human cytomegalovirus reduces the inhibitory effect of gH-specific and gB-specific antibodies. Arch. Virol. 2011, 156, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Hirt, B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967, 26, 365–369. [Google Scholar] [CrossRef]
- Park, J.; Gill, K.S.; Aghajani, A.A.; Heredia, J.D.; Choi, H.; Oberstein, A.; Procko, E. Engineered receptors for human cytomegalovirus that are orthogonal to normal human biology. PLoS Pathog. 2020, 16, e1008647. [Google Scholar] [CrossRef]
- Stegmann, C.; Rothemund, F.; Laib Sampaio, K.; Adler, B.; Sinzger, C. The N Terminus of Human Cytomegalovirus Glycoprotein O Is Important for Binding to the Cellular Receptor PDGFRα. J. Virol. 2019, 93, e00138-19. [Google Scholar] [CrossRef] [Green Version]
- Kschonsak, M.; Rougé, L.; Arthur, C.P.; Hoangdung, H.; Patel, N.; Kim, I.; Johnson, M.C.; Kraft, E.; Rohou, A.L.; Gill, A.; et al. Structures of HCMV Trimer reveal the basis for receptor recognition and cell entry. Cell 2021, 184, 1232–1244.e16. [Google Scholar] [CrossRef]
- Wu, K.; Oberstein, A.; Wang, W.; Shenk, T. Role of PDGF receptor-α during human cytomegalovirus entry into fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, E9889–E9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [Green Version]
- Wussow, F.; Chiuppesi, F.; Contreras, H.; Diamond, D.J. Neutralization of Human Cytomegalovirus Entry into Fibroblasts and Epithelial Cells. Vaccines 2017, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Heim, K.P.; Che, Y.; Chi, X.; Qiu, X.; Han, S.; Dormitzer, P.R.; Yang, X. Prefusion structure of human cytomegalovirus glycoprotein B and structural basis for membrane fusion. Sci. Adv. 2021, 7, eabf3178. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) |
|---|---|
| UL74-S48Y for | TTCTAACTTAAGCTGATCTAATTTATATTTGCCTATCTTATACAGTACCAAGCCCCTCCAAaggatgacgacgataagt |
| UL74-S48Y rev | AAAGCGCTTTATAATCGTCCTTGGAGGGGCTTGGTACTGTATAAGATAGGCAAATATAAATcaaccaattaaccaattctga |
| UL74-S48Y short for | TTCTAACTTAAGCTGATCTA |
| UL55-H177N for | CTGTAGGAACTGTAGCATTGAGCAAACTTGTTGATGTGATTAATCTCCCACATAGGAGGCGaggatgacgacgataagt |
| UL55-H177N rev | CAGCAATACGGAATACGTGGCGCCTCCTATGTGGGAGATTAATCACATCAACAAGTTTGCTcaaccaattaaccaattctga |
| UL55-H177N short for | CTGTAGGAACTGTAGCATTG |
| UL73-D84Y for | CACCCACGATCCTAATGTGATGAGACGACATGCGAACGATTATTTTTACAAGGCGCATTGCaggatgacgacgataagt |
| UL73-D84Y rev | AGCTCATACATATGCGATGTGCAATGCGCCTTGTAAAAATAATCGTTCGCATGTCGTCTCAcaaccaattaaccaattctga |
| UL73-D84Y short for | CACCCACGATCCTAATGTGA |
| UL74-L47P for | TAACTTAAGCTGATCTAATTTATATTTGCCTATCTTAGACGGTACCAAGCCCCTCCAAGGAaggatgacgacgataagt |
| UL74-L47P rev | TCAAAAGCGCTTTATAATCGTCCTTGGAGGGGCTTGGTACCGTCTAAGATAGGCAAATATAcaaccaattaaccaattctga |
| UL74-L47P short for | TAACTTAAGCTGATCTAATT |
| UL75-I417V for | TGTTTAGAAAGTATGTAGACCAGGCGTACGAGGCTGGTGACGTCGGTGATCTGGTCCGGCGaggatgacgacgataagt |
| UL75-I417V rev | GGCCAAACGAGCCCTCTGGACGCCGGACCAGATCACCGACGTCACCAGCCTCGTACGCCTGcaaccaattaaccaattctga |
| UL75-I417V for short | TGTTTAGAAAGTATGTAGAC |
| Kanamycin universal reverse | CAACCAATTAACCAATTCTGA |
| Primer | Sequence (5′-3′) |
|---|---|
| EGFP for | CCGTGCAGAACATCCTCCAAAAGATCGAGAAGATTAAGAAAACGGAGGAAatggtgagcaagggcgaggagct |
| EGFP rev | CACTATCCGATGATTTCATTAAAAAGTACGTCTGCGTGTGTGTTTCTTAAttacttgtacagctcgtccatgc |
| mCherry for | CCCTGCGTCTACTATCACGTCGTGGACTTTGAAAGGCTCAACATGTCGGCCTACAACGTAgtgagcaagggcgaggagga |
| mCherry rev | CACGGCGTAGCACACCAGCTGCACCGAGTCTAAGAAAAGCATAGGCGTGTGCAGGTGCATcttgtacagctcgtccatgc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laib Sampaio, K.; Lutz, C.; Engels, R.; Stöhr, D.; Sinzger, C. Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors. Viruses 2021, 13, 1094. https://doi.org/10.3390/v13061094
Laib Sampaio K, Lutz C, Engels R, Stöhr D, Sinzger C. Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors. Viruses. 2021; 13(6):1094. https://doi.org/10.3390/v13061094
Chicago/Turabian StyleLaib Sampaio, Kerstin, Carolin Lutz, Rebecca Engels, Dagmar Stöhr, and Christian Sinzger. 2021. "Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors" Viruses 13, no. 6: 1094. https://doi.org/10.3390/v13061094
APA StyleLaib Sampaio, K., Lutz, C., Engels, R., Stöhr, D., & Sinzger, C. (2021). Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors. Viruses, 13(6), 1094. https://doi.org/10.3390/v13061094