
PhaLP: A Database for the Study of Phage Lytic Proteins and Their Evolution

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
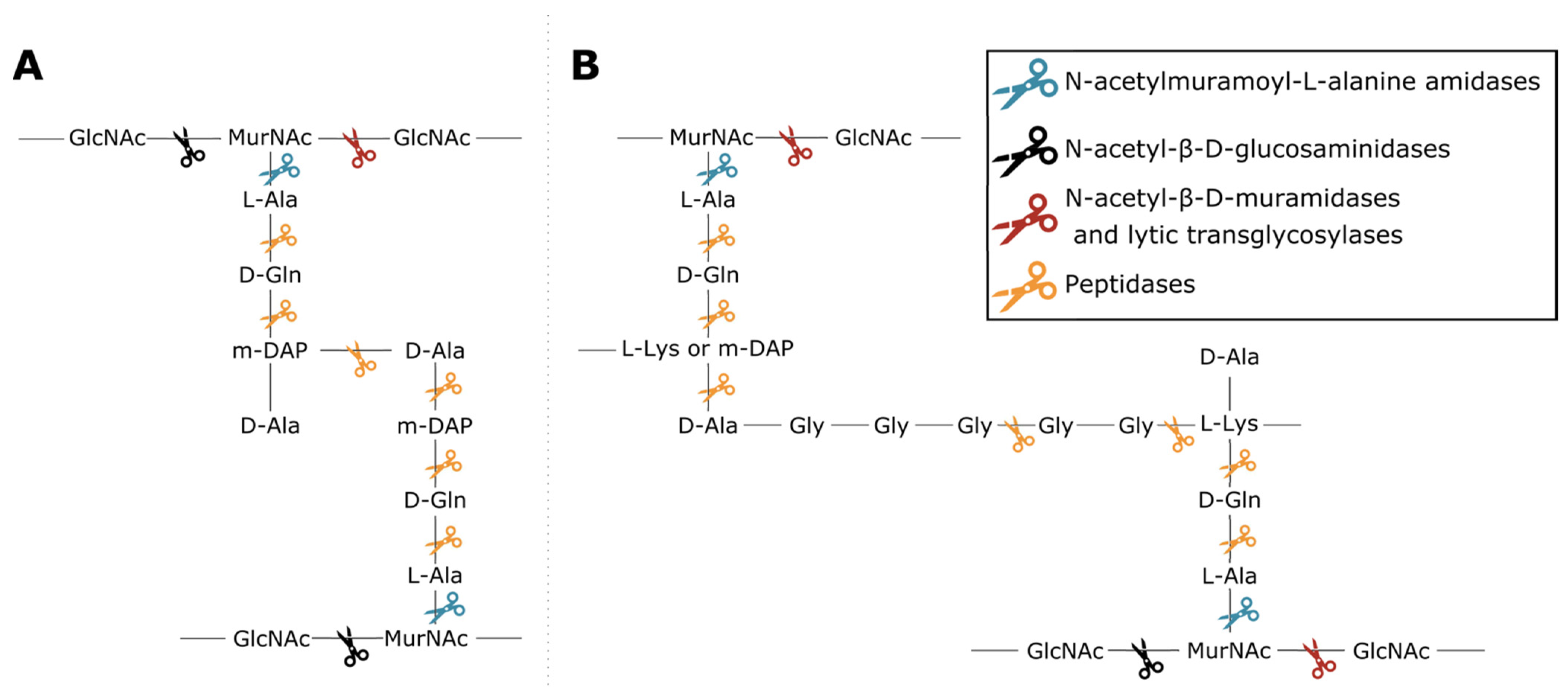
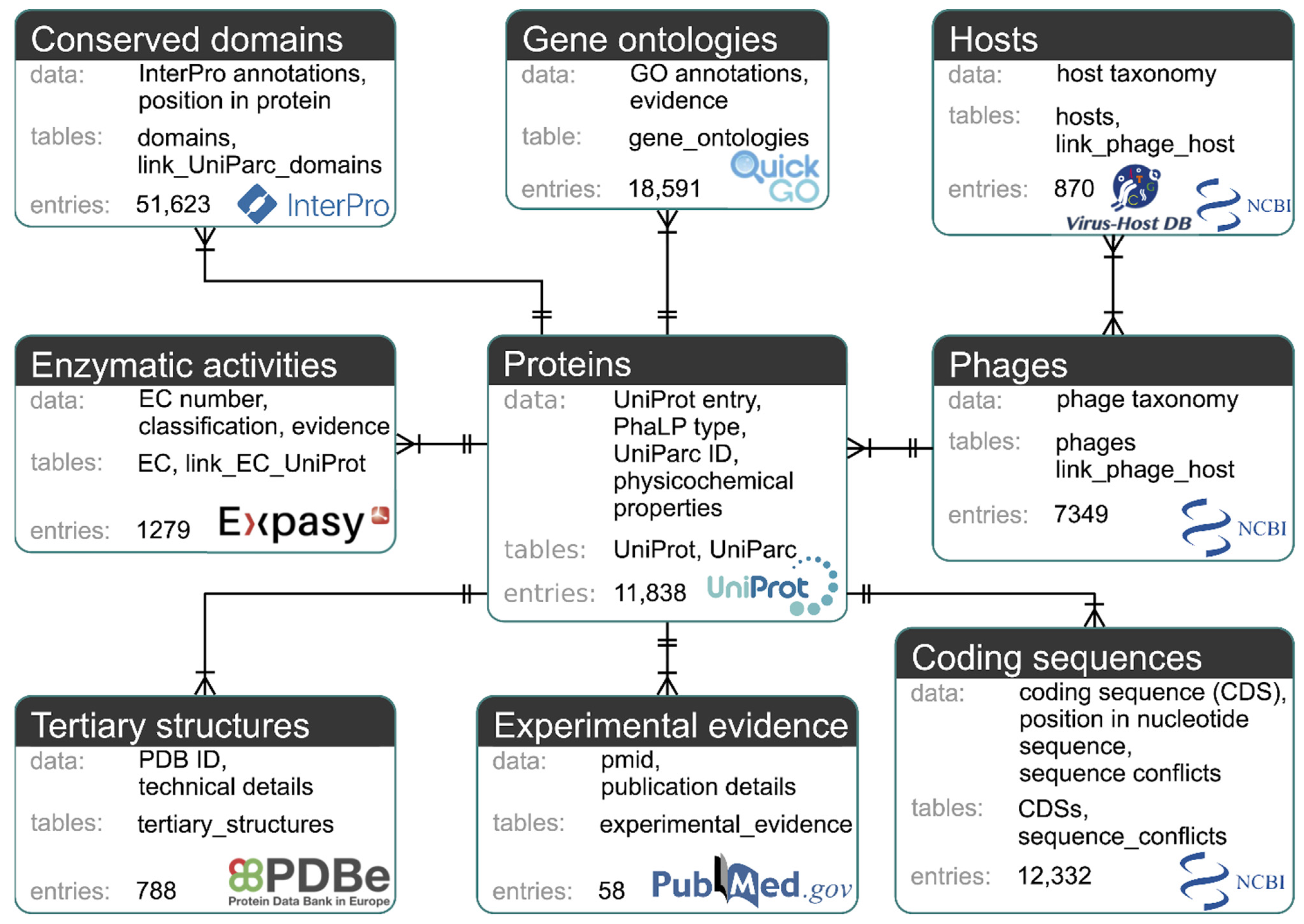
2.1. Database Structure and Construction
2.2. Type Classification
2.3. Accessing Data
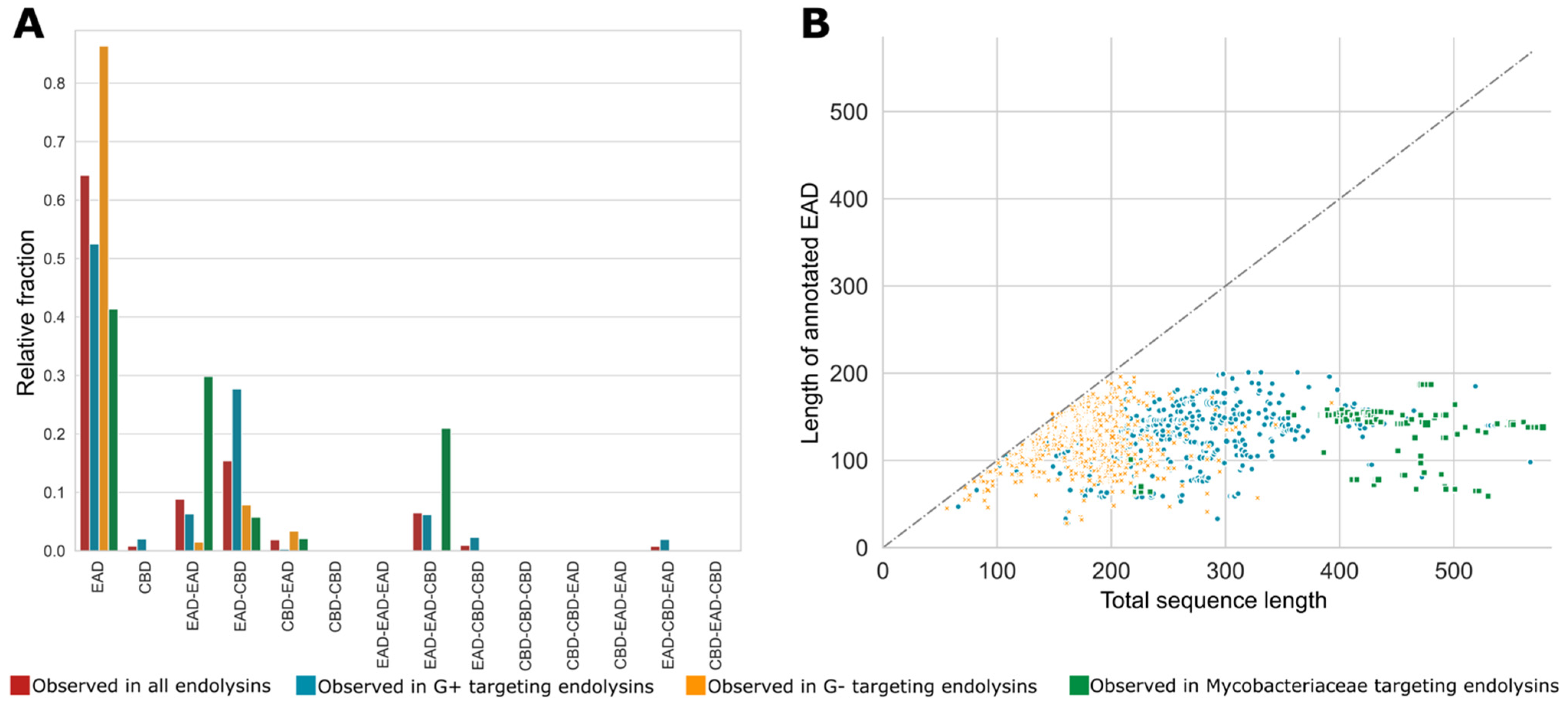
2.4. Quantitative Exploration of the Modular Composition
2.5. Host-Specific Evolution of Phage Lytic Proteins by Recombination
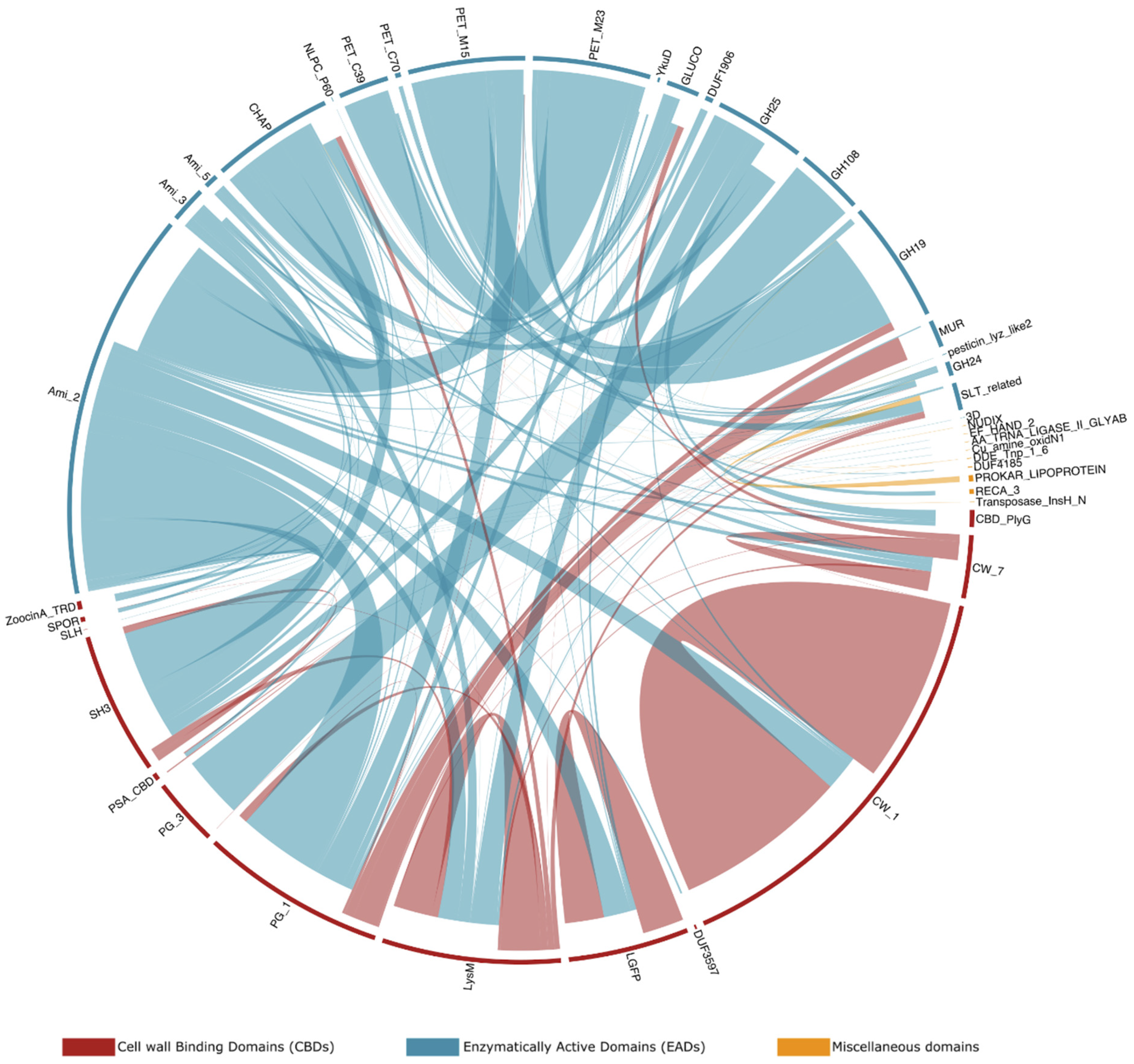
2.6. Modular Organization: Architectures and Adjacency
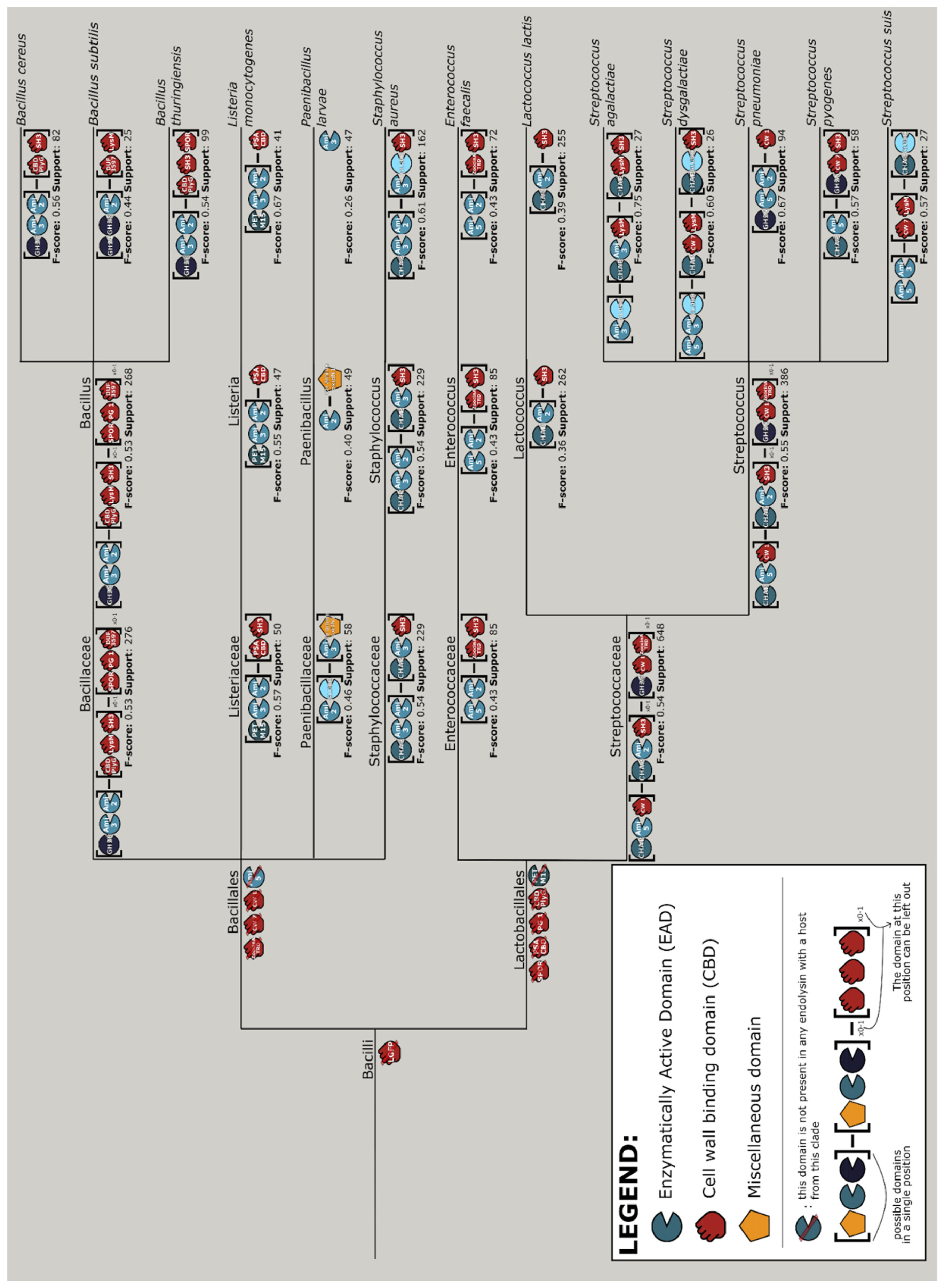
2.7. Aggregating Differences in Modular Compositions and Orders into Nature’s Design Rules
2.7.1. Machine Learning Approach
2.7.2. Data Mining Approach
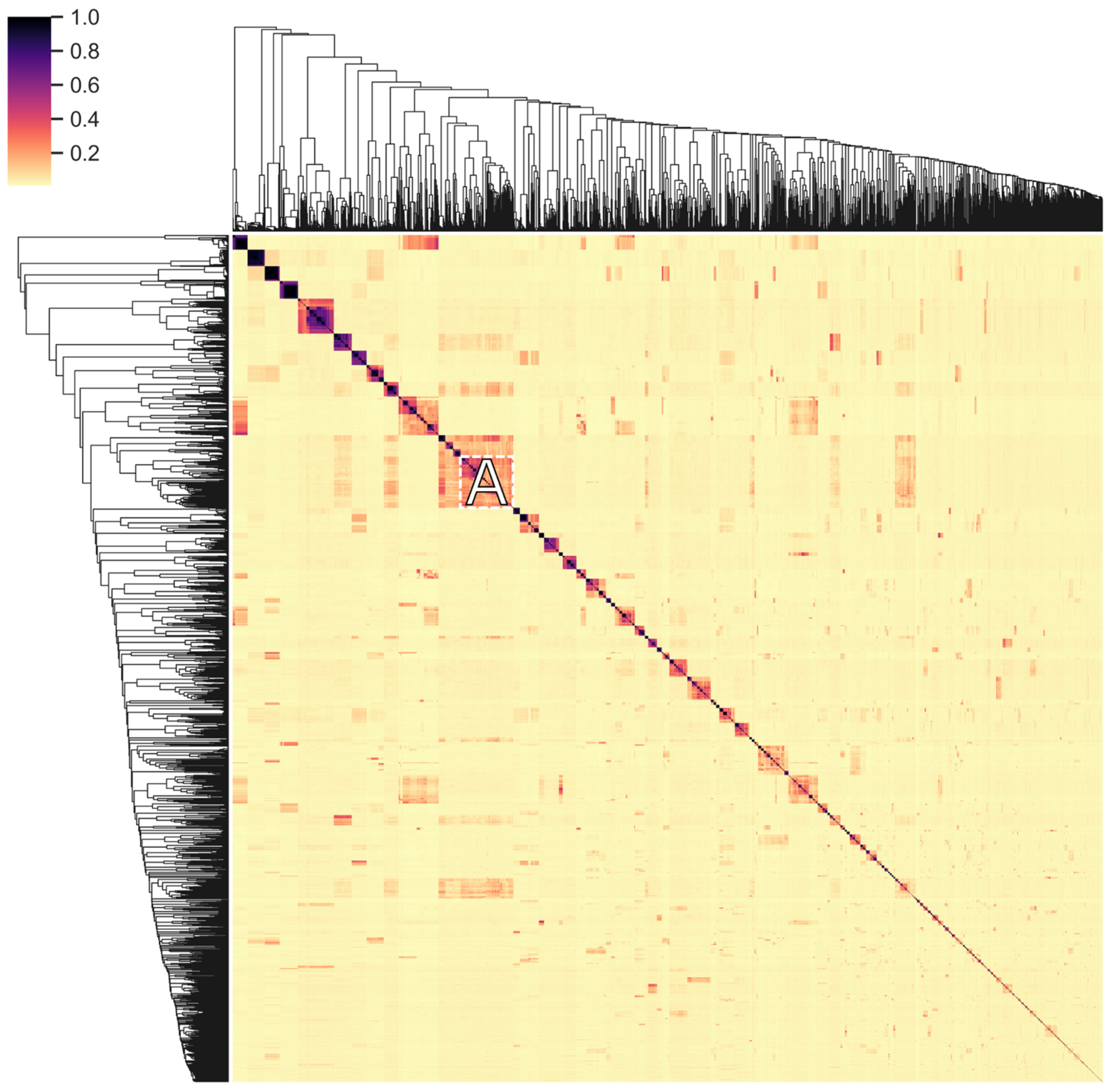
2.8. Vertical Evolution of Endolysins
3. Results and Discussion
3.1. Database Structure and Construction
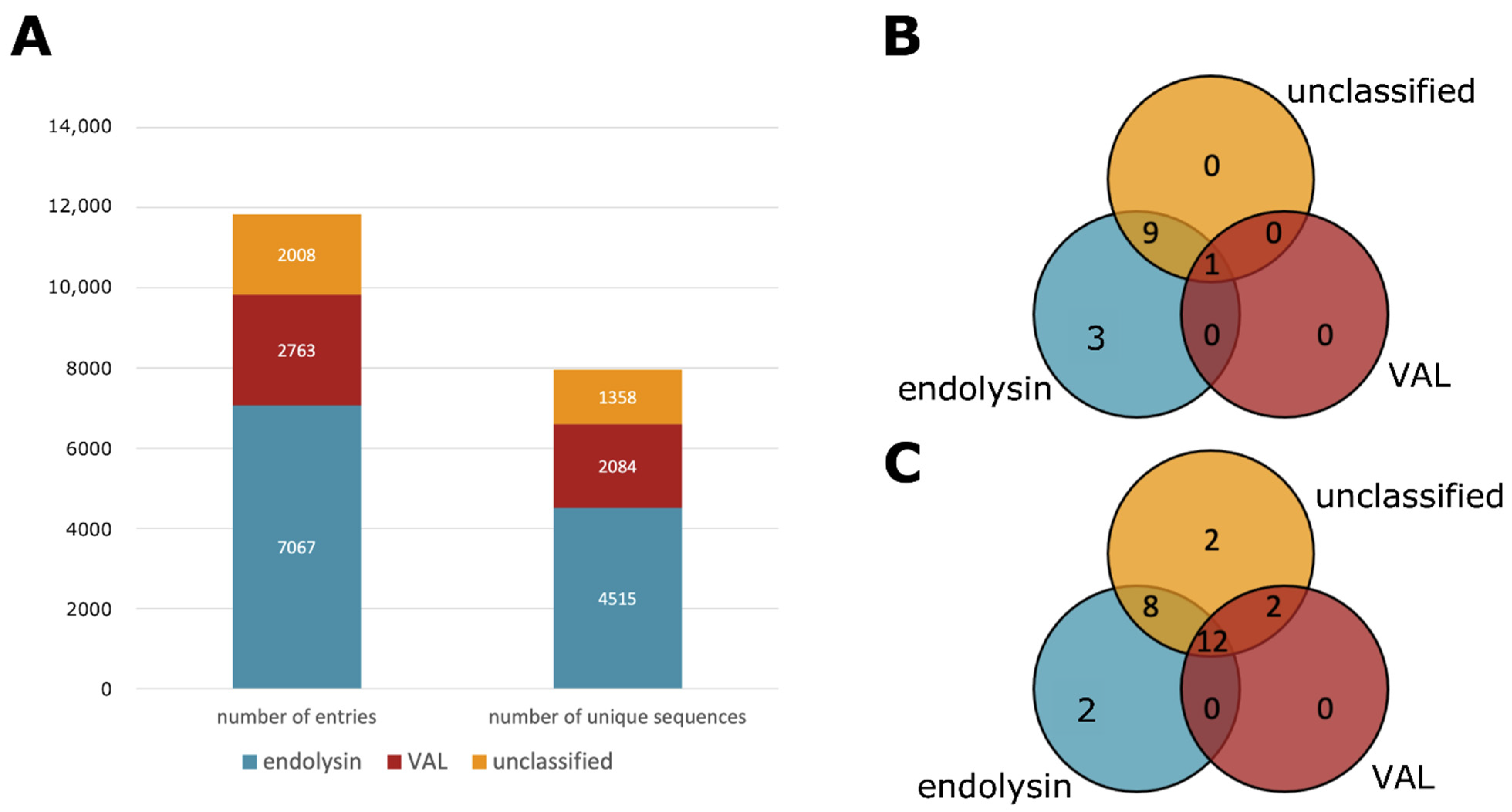
3.2. Type Classification
3.3. Accessing Data
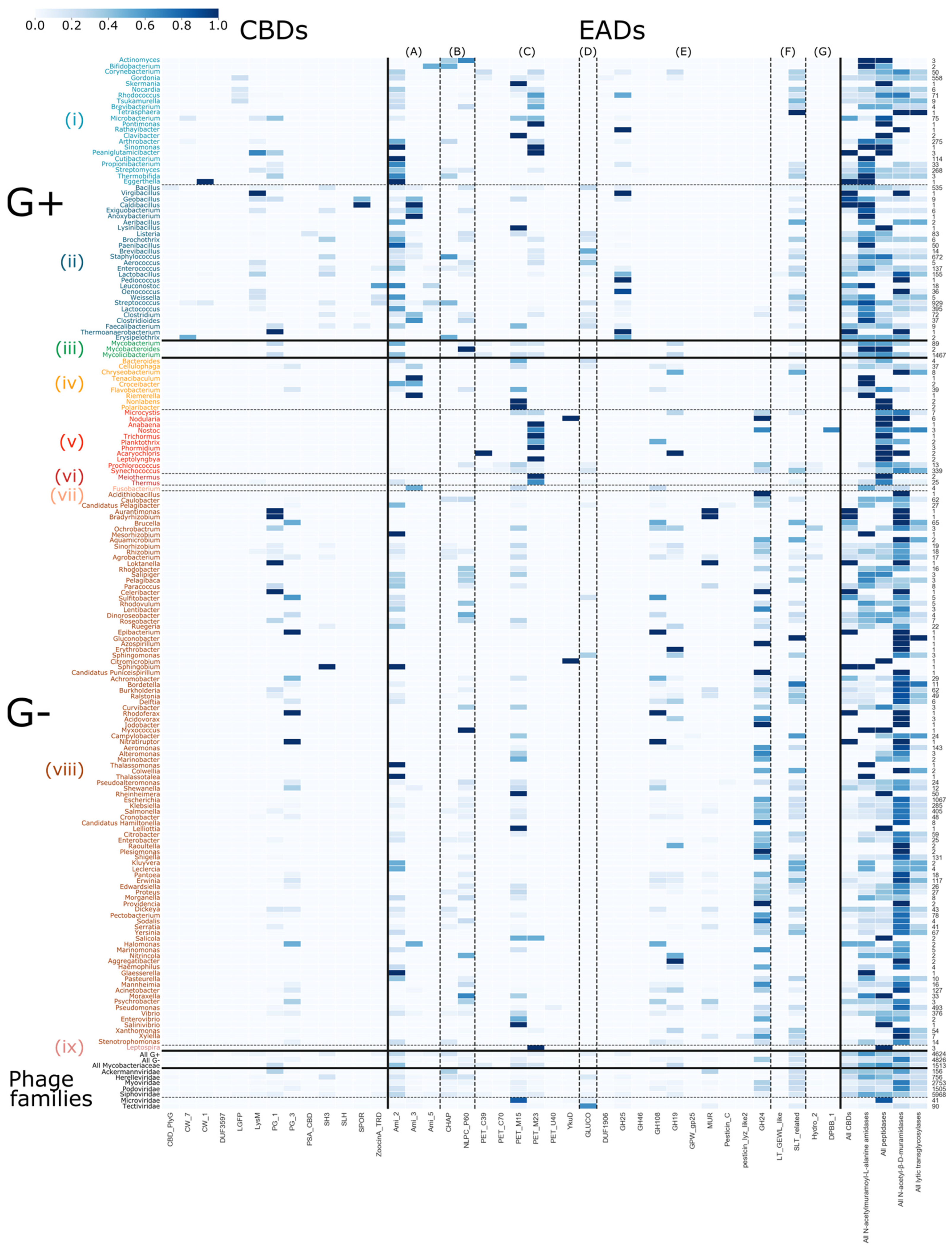
3.4. Quantitative Exploration of the Modular Composition
3.5. Host-Specific Evolution of Phage Lytic Proteins by Recombination
3.6. Modular Organization: Architectures and Adjacency
3.7. Aggregating Differences in Modular Compositions and Orders into Nature’s Design Rules
3.8. Vertical Evolution of Endolysins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandes, S.; São-José, C. Enzymes and Mechanisms Employed by Tailed Bacteriophages to Breach the Bacterial Cell Barriers. Viruses 2018, 10, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, D.; Fernández, L.; Rodríguez, A.; García, P. Are phage lytic proteins the secret weapon to kill Staphylococcus aureus? MBio 2018, 9, e01923-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-derived peptidoglycan degrading enzymes: Challenges and future prospects for in vivo therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [Green Version]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [CrossRef]
- Catalão, M.J.; Gil, F.; Moniz-Pereira, J.; São-José, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. FEMS Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Loessner, M.J.; Kramer, K.; Ebel, F.; Scherer, S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol. Microbiol. 2002, 44, 335–349. [Google Scholar] [CrossRef]
- São-José, C. Engineering of phage-derived lytic enzymes: Improving their potential as antimicrobials. Antibiotics 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briers, Y.; Volckaert, G.; Cornelissen, A.; Lagaert, S.; Michiels, C.W.; Hertveldt, K.; Lavigne, R. Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages φKZ and EL. Mol. Microbiol. 2007, 65, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol. Adv. 2018, 36, 624–640. [Google Scholar] [CrossRef]
- Loessner, M.J. Bacteriophage endolysins—Current state of research and applications. Curr. Opin. Microbiol. 2005, 8, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, N.; Campillo, N.E.; García, E.; Gallego, C.; Pera, B.; Diakun, G.P.; Sáiz, J.L.; García, P.; Díaz, J.F.; Menéndez, M. Cpl-7, a lysozyme encoded by a pneumococcal bacteriophage with a novel cell wall-binding motif. J. Biol. Chem. 2010, 285, 33184–33196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell wall hydrolases in bacteria: Insight on the diversity of cell wall amidases, glycosidases and peptidases toward peptidoglycan. Front. Microbiol. 2019, 10, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Eugster, M.R.; Haug, M.C.; Huwiler, S.G.; Loessner, M.J. The cell wall binding domain of Listeria bacteriophage endolysin PlyP35 recognizes terminal GlcNAc residues in cell wall teichoic acid. Mol. Microbiol. 2011, 81, 1419–1432. [Google Scholar] [CrossRef]
- Oliveira, H.; Melo, L.D.R.; Santos, S.B.; Nobrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yu, J.; Wei, H. Engineered bacteriophage lysins as novel anti-infectives. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Maxted, W.R. The active agent in nascent phage lysis of streptococci. J. Gen. Microbiol. 1957, 16, 584–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, R.M. Studies on bacteriophages of hemolytic streptococci. I. Factors influencing the interaction of phage and susceptible host cell. J. Exp. Med. 1957, 106, 365–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [Green Version]
- Heselpoth, R.D.; Euler, C.W.; Schuch, R.; Fischetti, V.A. Lysocins: Bioengineered antimicrobials that deliver lysins across the outer membrane of Gram-negative bacteria. Antimicrob. Agents Chemother. 2019, 63, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampara, A.; Sørensen, M.C.H.; Grimon, D.; Antenucci, F.; Vitt, A.R.; Bortolaia, V.; Briers, Y.; Brøndsted, L. Exploiting phage receptor binding proteins to enable endolysins to kill Gram-negative bacteria. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Grishin, A.V.; Karyagina, A.S.; Vasina, D.V.; Vasina, I.V.; Gushchin, V.A.; Lunin, V.G. Resistance to peptidoglycan-degrading enzymes. Crit. Rev. Microbiol. 2020, 46, 703–726. [Google Scholar] [CrossRef] [PubMed]
- Hojckova, K.; Stano, M.; Klucar, L. PhiBIOTICS: Catalogue of therapeutic enzybiotics, relevant research studies and practical applications. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Lu, H.; Huang, J.; Li, G.; Huang, Q. EnzyBase: A novel database for enzybiotic studies. BMC Microbiol. 2012, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinform. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Django. 2019. Available online: https://www.djangoproject.com (accessed on 25 June 2021).
- Zhang, J.; Haider, S.; Baran, J.; Cros, A.; Guberman, J.M.; Hsu, J.; Liang, Y.; Yao, L.; Kasprzyk, A. BioMart: A data federation framework for large collaborative projects. Database 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M. The Seaborn Development Team Mwaskom/Seaborn. 2020. Available online: https://doi.org/10.5281/zenodo.592845 (accessed on 25 June 2021).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Gardin Gautier, G.N.S. Skope Rules: Machine Learning with Logical Rules in Python. 2017. Available online: https://github.com/scikit-learn-contrib/skope-rules (accessed on 25 June 2021).
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking virus genomes with host taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rubio, L.; Martínez, B.; Rodríguez, A.; Donovan, D.M.; Götz, F.; García, P. The phage lytic proteins from the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 display multiple active catalytic domains and do not trigger staphylococcal resistance. PLoS ONE 2013, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; Rodríguez, A.; García, P. Bacteriophage virion-associated peptidoglycan hydrolases: Potential new enzybiotics. Crit. Rev. Microbiol. 2013, 39, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.L.; Hassanabad, M.F.; Chang, T.; Pirani, N.; Bona, D.; Edwards, A.M.; Davidson, A.R. Structural and functional studies of gpX of Escherichia coli phage P2 reveal a widespread role for LysM domains in the baseplates of contractile-tailed phages. J. Bacteriol. 2013, 195, 5461–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelcher, M.; Tchang, V.S.; Loessner, M.J. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb. Biotechnol. 2011, 4, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kwon, S.-J.; Sauve, J.; Fraser, K.; Kemp, L.; Lee, I.; Nam, J.; Kim, J.; Dordick, J.S. Modular Assembly of Unique Chimeric Lytic Enzymes on a Protein Scaffold Possessing Anti-Staphylococcal Activity. Biomacromolecules 2019, 20, 4035–4043. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Grimon, D.; Gutiérrez, D.; Lood, C.; Rodríguez, A.; van Noort, V.; Lammertyn, J.; Lavigne, R.; Briers, Y. A VersaTile-driven platform for rapid hit-to-lead development of engineered lysins. Sci. Adv. 2020, 6, eaaz1136. [Google Scholar] [CrossRef]
- Duyvejonck, L.; Gerstmans, H.; Stock, M.; Grimon, D.; Lavigne, R.; Briers, Y. Rapid and High-Throughput Evaluation of Diverse Configurations of Engineered Lysins Using the VersaTile Technique. Antibiotics 2021, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Ryu, S. Characterization of a novel cell wall binding domain-containing Staphylococcus aureus endolysin LysSA97. Appl. Microbiol. Biotechnol. 2017, 101, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Oliveira, A.; Melo, L.D.R.; Azeredo, J. Identification of the first endolysin Cell Binding Domain (CBD) targeting Paenibacillus larvae. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.; Schuch, R.; Chahales, P.; Zhu, S.; Fischetti, V.A. PlyC: A multimeric bacteriophage lysin. Proc. Natl. Acad. Sci. USA 2006, 103, 10765–10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoiczyk, E.; Hansel, A. Cyanobacterial cell walls: News from an unusual prokaryotic envelope. J. Bacteriol. 2000, 182, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, A.M.; Hampton, C.M.; Dillard, R.S.; Gil, F.; Catalão, M.J.; Moniz-Pereira, J.; Wright, E.R.; Pimentel, M. The Ms6 mycolyl-arabinogalactan esterase lysB is essential for an efficient mycobacteriophage-induced lysis. Viruses 2017, 9, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, J.M.; Duffy, S.; Sanjuán, R. Point mutation rate of bacteriophage ΦX174. Genetics 2009, 183, 747–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. BioEssays 2011, 33, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymatic Activity a | Domain Cluster Name b | Occurrences in PhaLP c | Domain Accessions (Occurrences in PhaLP) d |
|---|---|---|---|
| (A) | Ami_2 | 2309 | SM00644 (2078), SM00701 (302), cd06583 (1923), PF01510 (2291) |
| Ami_3 | 294 | SM00646 (269), cd02696 (290), PF01520 (294) | |
| Ami_5 | 314 | PF05382 (314) | |
| (B) | CHAP | 1127 | PF05257 (955), PS50911 (958) |
| NLPC_P60 | 747 | PF00877 (747) | |
| (C) | PET_M15 | 1081 | cd14814 (103), cd14852 (2), PF02557 (55), cd14849 (1), cd14844 (210), PF08291 (248), cd14845 (632), PF13539 (724) |
| PET_M23 | 782 | PF01551 (782) | |
| PET_C39 | 260 | PF13529 (260) | |
| PET_U40 | 38 | PF10464 (38) | |
| PET_C70 | 19 | PF12385 (19) | |
| YkuD | 10 | PF10908 (1), cd16913 (10), PF03734 (4) | |
| (D) | GLUCO | 541 | PF01832 (541), SM00047 (504) |
| (E) | GH24 | 1834 | cd00737 (466), cd00736 (181), cd16900 (424), cd16901 (121), PF00959 (1546), cd00735 (642) |
| GH19 | 748 | cd00325 (743), PF00182 (283) | |
| GH25 | 388 | cd00599 (90), cd06417 (1), cd06415 (83), cd06414 (4), cd06525 (3), PF01183 (388), SM00641 (304), cd06522 (1), cd06523 (32) | |
| GH108 | 254 | cd13926 (254), PF05838 (250) | |
| MUR | 162 | PF11860 (162) | |
| DUF1906 | 30 | PF08924 (30) | |
| GH46 | 10 | cd00978 (10), PF01374 (10) | |
| Pesticin_C | 6 | cd16902 (6), PF16754 (6) | |
| GPW_gp25 | 1 | PF04965 (1) | |
| Pesticin_lyz_like2 | 1 | cd16904 (1) | |
| (F) | SLT_related | 2202 | PS51348 (1), cd16899 (16), PF00062 (1), cd13399 (21), cd16896 (17), PF01464 (1138), cd00254 (517), cd13401 (52), cd01021 (33), cd16893 (21), cd16894 (101), cd13400 (94), cd13403 (79), cd13925 (273), PF06737 (203), cd13402 (529), PF18013 (329) |
| LT_GEWL_like | 24 | cd16891 (24), PF13702 (24) | |
| (G) | Hydro_2 | 32 | PF07486 (32) |
| DPBB_1 | 2 | PF03330 (2) |
| Domain Cluster Name a | Occurrences in PhaLP b | Domain Accessions (Occurrences in PhaLP) c |
|---|---|---|
| CBD_PlyG | 54 | PF12123 (54) |
| CW_7 | 186 | PF08230 (186), SM01095 (186) |
| CW_1 | 757 | PF01473 (628), PS51170 (757) |
| DUF3597 | 8 | PF12200 (8) |
| LGFP | 276 | PF08310 (276) |
| LysM | 427 | PF01476 (422), cd00118 (425), PS51782 (425), SM00257 (415) |
| PG_1 | 640 | PF01471 (640) |
| PG_3 | 235 | PF09374 (235) |
| PSA_CBD | 18 | PF18341 (18) |
| SH3 | 552 | PF08239 (34), PF08460 (462), PS51781 (179), SM00287 (414) |
| SLH | 2 | PS51272 (1), PF00395 (1) |
| SPOR | 22 | PF05036 (21), PS51724 (17) |
| ZoocinA_TRD | 250 | PF16775 (250) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criel, B.; Taelman, S.; Van Criekinge, W.; Stock, M.; Briers, Y. PhaLP: A Database for the Study of Phage Lytic Proteins and Their Evolution. Viruses 2021, 13, 1240. https://doi.org/10.3390/v13071240
Criel B, Taelman S, Van Criekinge W, Stock M, Briers Y. PhaLP: A Database for the Study of Phage Lytic Proteins and Their Evolution. Viruses. 2021; 13(7):1240. https://doi.org/10.3390/v13071240
Chicago/Turabian StyleCriel, Bjorn, Steff Taelman, Wim Van Criekinge, Michiel Stock, and Yves Briers. 2021. "PhaLP: A Database for the Study of Phage Lytic Proteins and Their Evolution" Viruses 13, no. 7: 1240. https://doi.org/10.3390/v13071240
APA StyleCriel, B., Taelman, S., Van Criekinge, W., Stock, M., & Briers, Y. (2021). PhaLP: A Database for the Study of Phage Lytic Proteins and Their Evolution. Viruses, 13(7), 1240. https://doi.org/10.3390/v13071240