
Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex



Abstract
:1. Introduction
2. Bacteria of the Burkholderia cepacia Complex
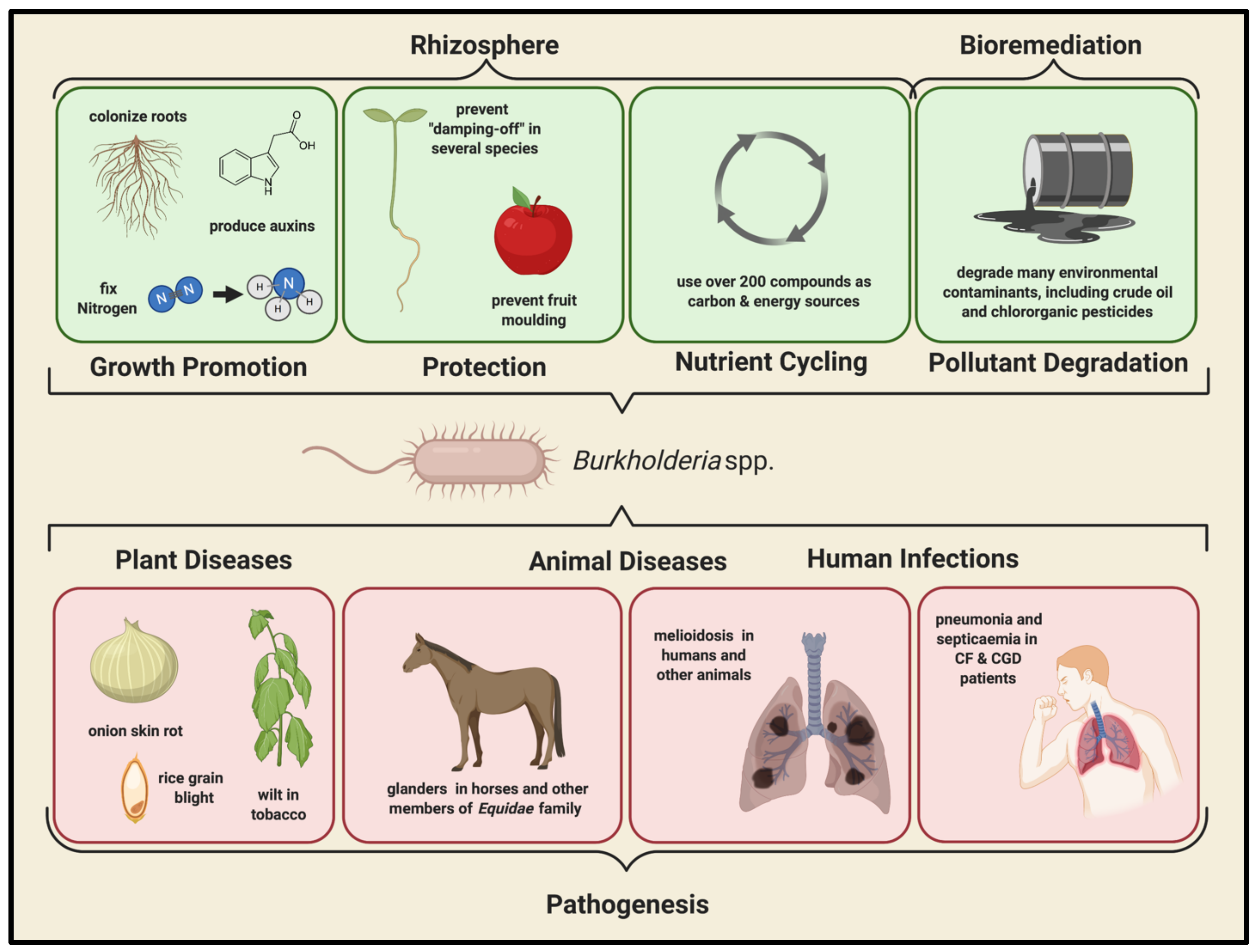
2.1. Environmental Importance of the Bcc
2.2. Clinical Importance of the Bcc
2.3. Bcc Infections
2.3.1. Cystic Fibrosis (CF)
2.3.2. Chronic Granulomatous Disease (CGD)
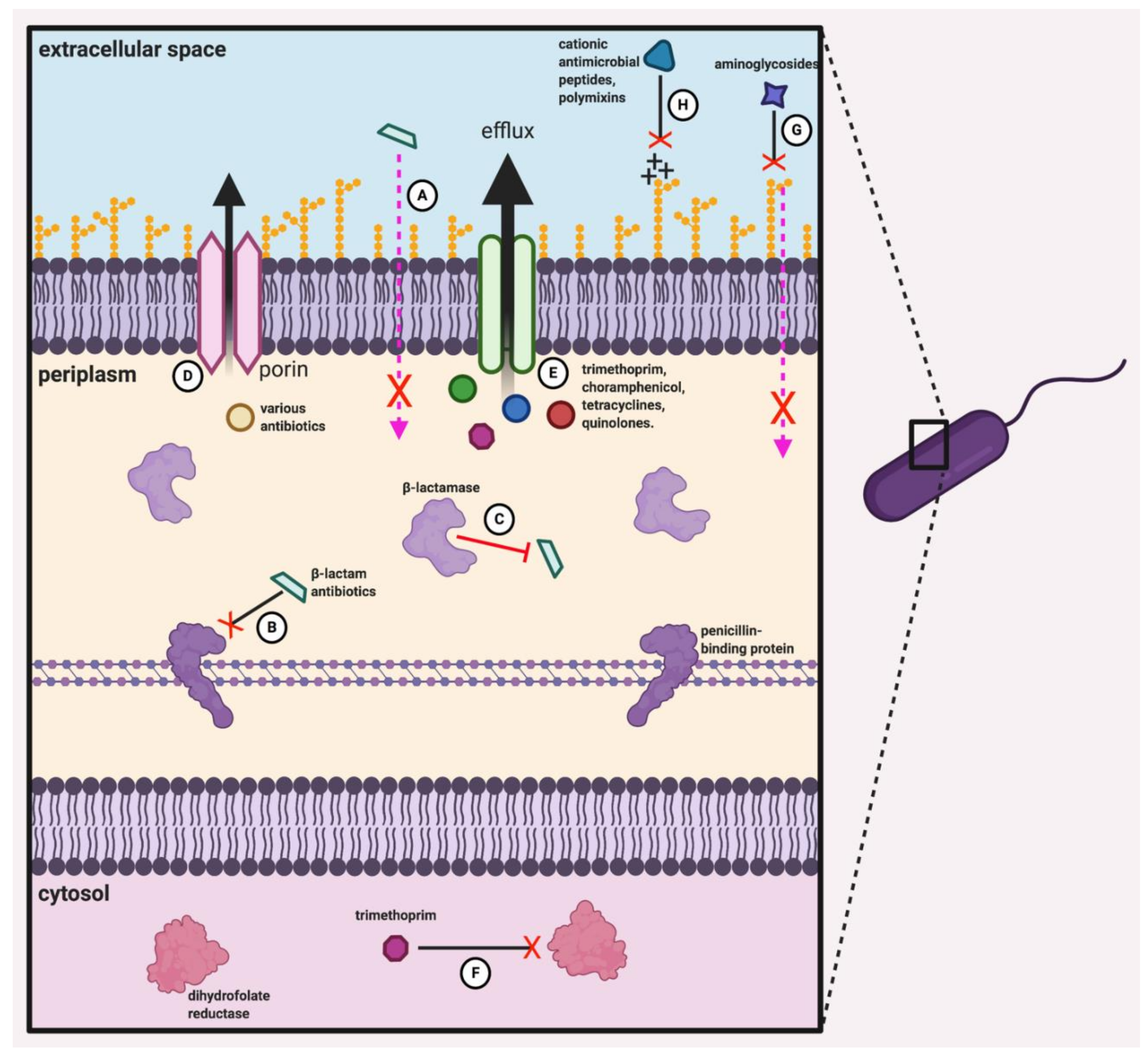
2.4. Antibiotic Resistance of the Bcc
3. Bacteriophage Therapy
3.1. Lessons from History: Phage Therapy in the 19th and 20th Centuries
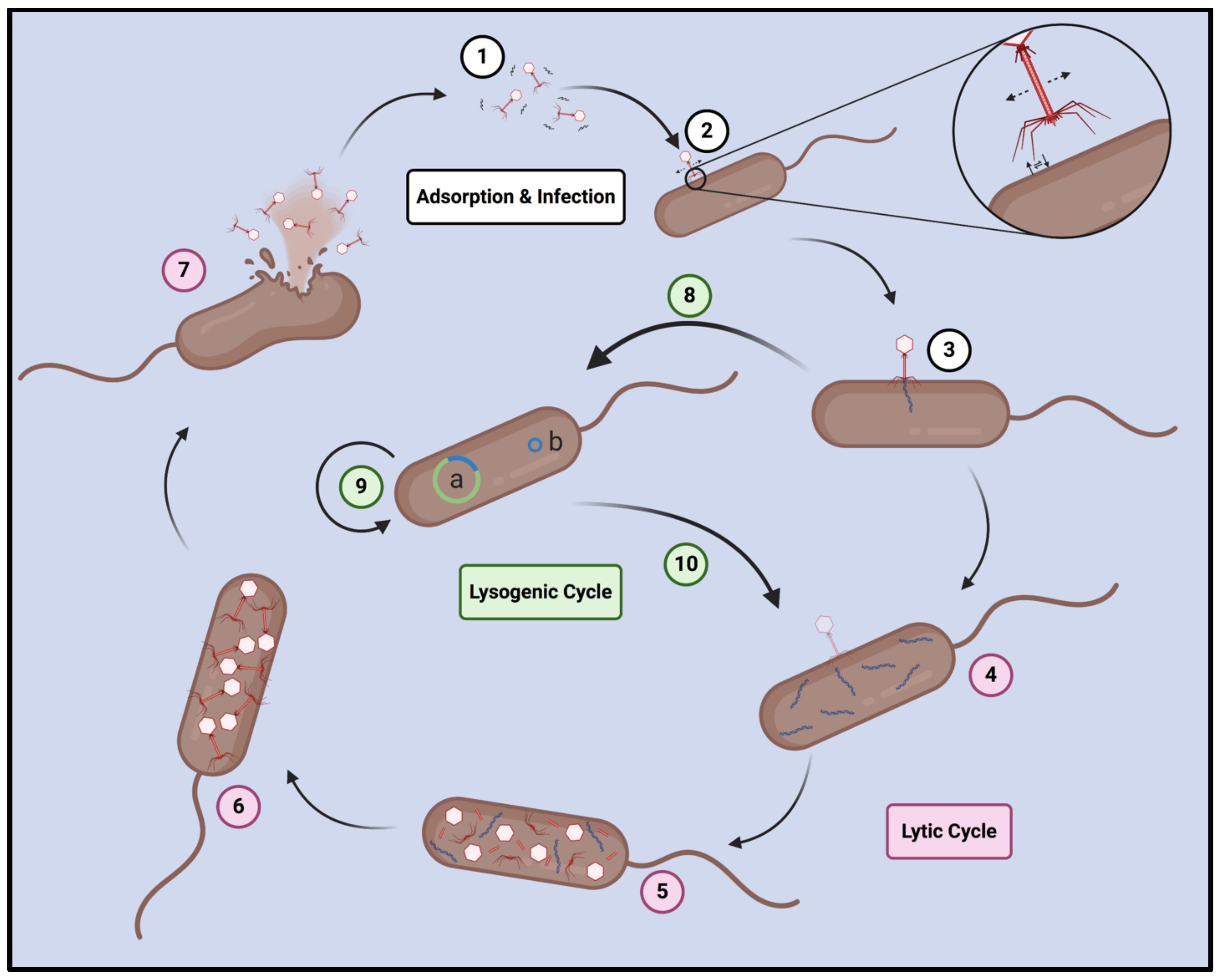
3.2. Bacteriophage Lifestyle and Mechanisms of Infection
3.3. Advantages of Phage Therapy Relative to Traditional Antibiotics
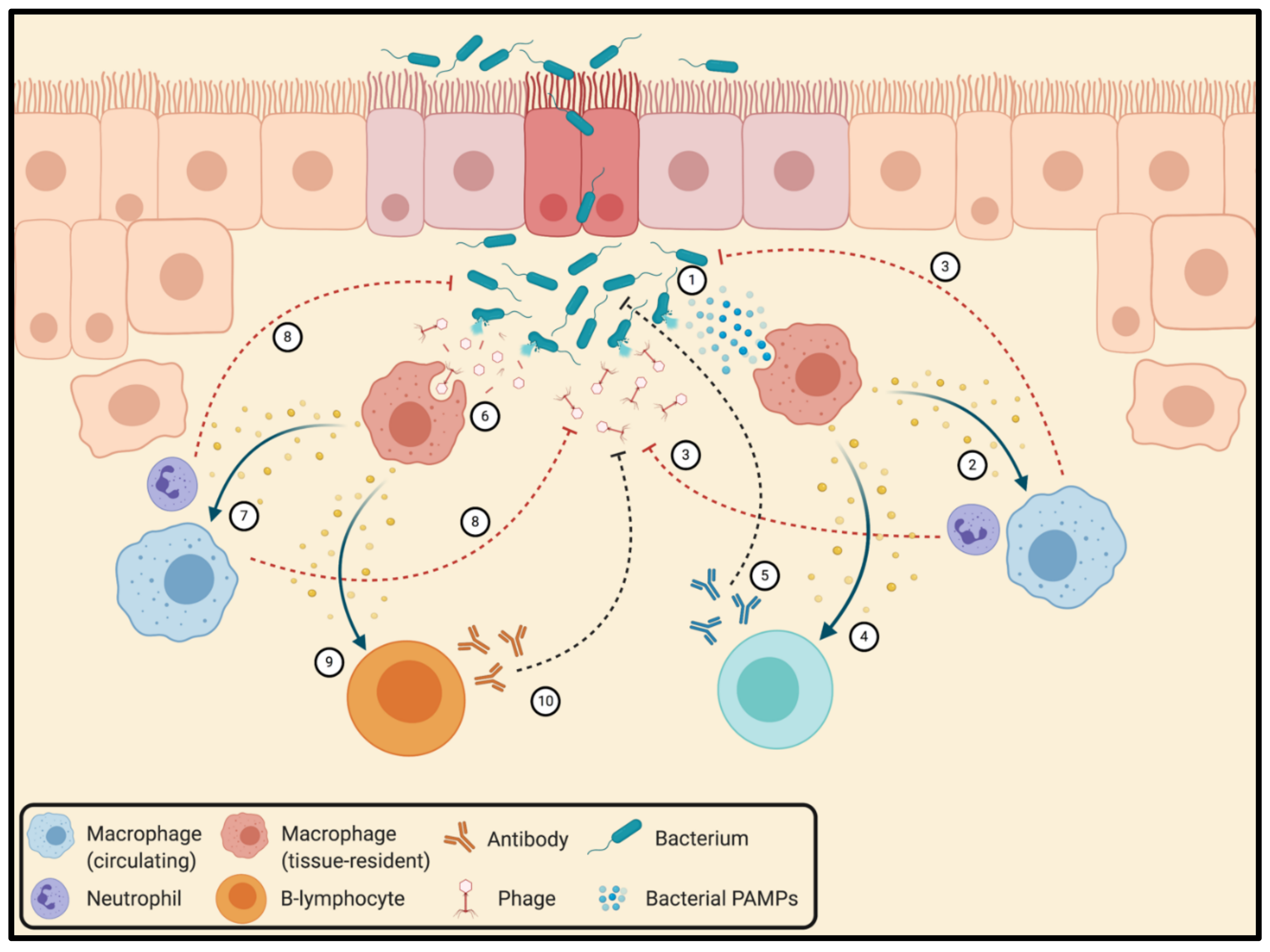
3.4. Interactions between Bacteriophages and the Mammalian Immune System
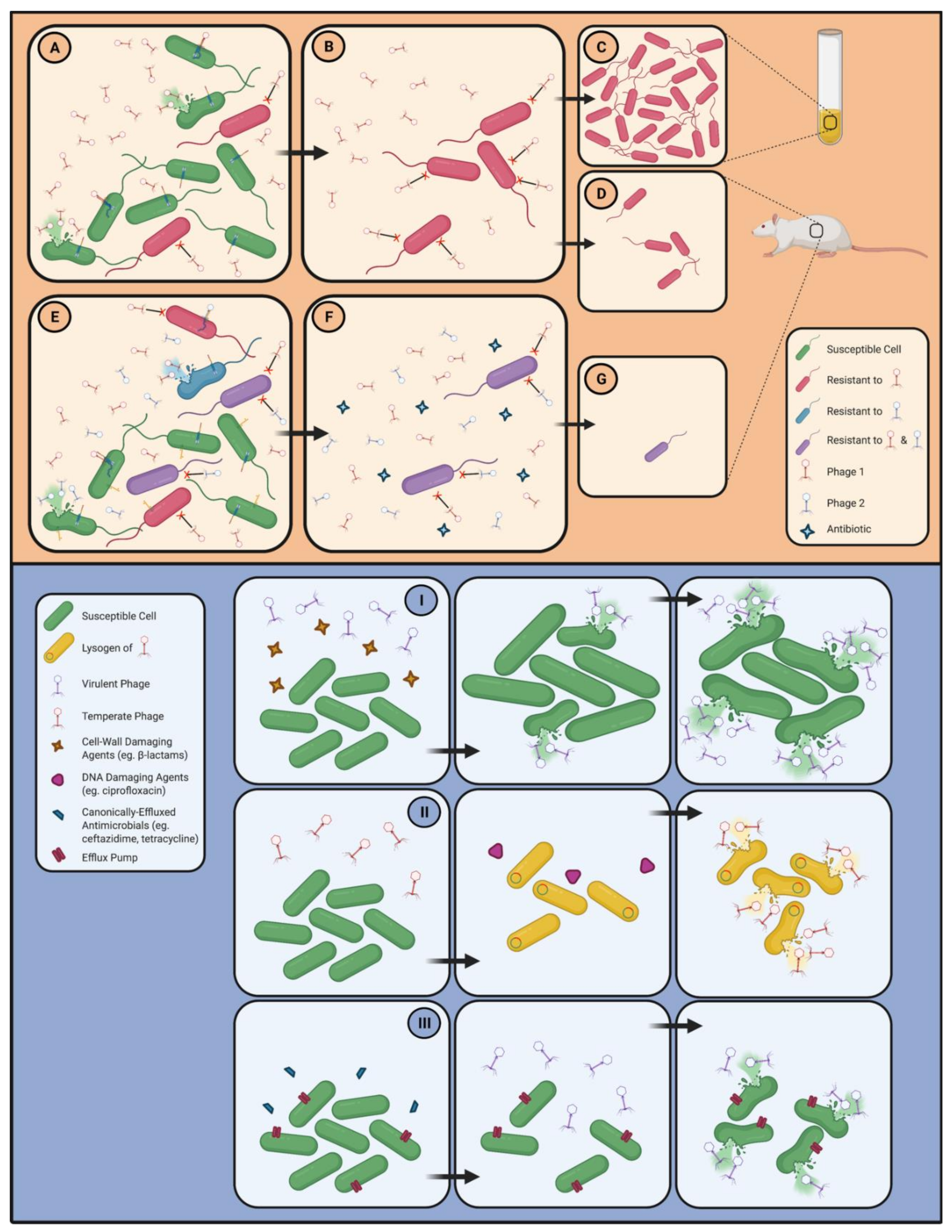
3.5. Strategies for the Circumvention of Problematic Phage Resistance
3.5.1. The Anti-Virulence Strategy
3.5.2. Polyphage Cocktails
3.5.3. Phage–Antibiotic Cocktails and Phage–Antibiotic Synergy
4. Bacteriophage Therapy Targeting the Burkholderia cepacia Complex
4.1. Bacteriophages Targeting the Bcc
4.2. Bcc Phage Therapy 2.0

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Name † | Morphology | Genus (Closest Relative) | Source | Bcc Host Range [# Known Hosts] | Lifestyle | Notes | ||
|---|---|---|---|---|---|---|---|---|
| General | Antibacterial Effects | |||||||
| Dennis Lab | KS1 [175] | Myoviridae | Unknown (not sequenced) | Onion rhizosphere | B. cenocepacia (715J, J2315, K56-2, C6433), B. ambifaria (LMG 19467) [7 hosts] | Unknown (not sequenced) | ||
| DK4/BcepMu/KS4 ‡ [175,176,255,263] | Myoviridae | Bcepmuvirus (Burkholderia phage | Lysogen of B. cenocepacia J2315 | B. cenocepacia (K56-2), B. ambifaria (LMG 19467) [2 hosts] | Lysogeny-capable | Unstable in lysate, but stable in G. mellonella haemolymph [169,175]. | ||
| KS4-M [175] | Myoviridae | Bcepmuvirus (Burkholderia phage | Mutant of DK4/BcepMu/KS4 | B. multivorans (C5393), B. cenocepacia (K56-2, C6433), B. ambifaria (LMG 19467) [4 hosts] | Lysogeny-capable | Stable in lysate (unlike parent phage) and in haemolymph. Can be aerosolized and nebulized for inhalatory delivery without significant loss of titer [169,175,264,269,270]. | Reduces mortality in G. mellonella. Produces a 2.5-log reduction in bacterial density in murine lung infection model [169,170]. | |
| KS5 [175] | Myoviridae | Kisquinquevirus (Burkholderia phage Mana) | Onion rhizosphere | B. cepacia (LMG 18821), B. multivorans (C5393), B. cenocepacia (715J, J2315, K56-2, C6433, C5424), B. ambifaria (LMG 19467) [8 hosts] | Lysogeny-capable; integrates at 3′ end of AMP nucleosidase [60]. | Primary receptor is LPS (lipid A proximal element is thought to be required) [60]. | Decreases bacterial density in murine lung infection model when applied at high MOI, but low MOI has little effect [170]. | |
| KS6 [175] | Myoviridae | Unknown (not sequenced) | Onion rhizosphere | B. cepacia (LMG 18821), B. multivorans (C5393, C5274), B. cenocepacia (715J, J2315, K56-2, C6433, C5424), B. stabilis (LMG 18870), B. ambifaria (LMG 19467) [10 hosts] | Unknown (not sequenced) | |||
| KS9 [175] | Siphoviridae | Unclassified Siphoviridiae (Burkholderia phage Bcep176) | Lysogen of B. pyrrocinia LMG 21824 | B. cenocepacia (K56-2), B. ambifaria (LMG 19467) [2 hosts] | Lysogeny-capable; integrates into 3′ end of GTP cyclohydrase II gene [166]. | Primary receptor is LPS (lipid A distal element required). Only Bcc phage that has been converted to obligately lytic form via insertional inactivation of lytic repressor [166]. | Reduces mortality in G. mellonella; effectiveness of obligately lytic form is not significantly different from that of the wild-type phage [166]. | |
| KS10 [175] | Myoviridae | Unclassified Myoviridae (Burkholderia phage BcepMu) | Lysogen of B. cenocepacia K56-2 | B. ambifaria (LMG 19467), B. cenocepacia (PC184), B. stabilis (LMG 18870) [3 hosts] | Lysogeny-capable; integrates [260]. | Transposable phage [260]. | ||
| KS14 [60] | Myoviridae | Kisquattuordecimvirus (Burkholderia phage FLC5) | Dracaena sp. soil | B. multivorans (C5393, C5274), B. cenocepacia (715J, C6433, C5424, PC184), B. dolosa (LMG 21443), B. ambifaria (LMG 17828) [8 hosts] | Lysogeny-capable; forms phagemid | Not stable in haemolymph (4-log titer loss in 24 h). Can be aerosolized for use in inhalatory delivery without significant loss of titer [169,270]. | Reduces mortality in G. mellonella, even at low MOIs. Produces significant reduction in bacterial density in murine lung infection model. Interacts synergistically with ciprofloxacin, meropenem and tetracycline based on plaque diameter [169,170,244]. | |
| KL3 [60] | Myoviridae | Tigrivirus (Burkholderia phage ) | Lysogen of B. cenocepacia CEP511 | B. ambifaria (LMG 17828) [1 host] | Lysogeny-capable; integrates into threonine tRNA gene | |||
| DC1 [271] | Podoviridae | Lessievirus (Burkholderia phage BcepIL02) | Dracaena sp. soil | B. cepacia (LMG 18821), B. cenocepacia (K56-2, C6433, PC184, CEP511), B. stabilis (LMG 18870) [6 hosts] | Lysogeny-capable; integrates; lysogens are unstable | Encodes a CsrA-like protein known to downregulate biofilm formation in E. coli. Stable in haemolymph [169,271]. | Fails to reduce mortality in G. mellonella at any MOI. Reduces bacterial density in murine lung infection model, but is the least efficient Bcc phage tested for this purpose [169,170]. | |
| KL1 [257] | Siphoviridae | Kilunavirus (Pseudomonas phage PaeS_SCUT-S4) | sewage | B. cenocepacia (K56-2, C6433, 715J, K63-3) [4 hosts] | Lysogeny-capable | Primary receptor is not LPS. Encodes MazG [257]. | ||
| AH2 [257] | Siphoviridae | Ahduovirus (Burkholderia phage BcepNazgul) | Nandina sp. soil | B. cenocepacia (K56-2, C6433, 715J, K63-3) [4 hosts] | Lysogeny-capable | Primary receptor is not LPS. Encodes MazG [257]. | ||
| KS12 [169] | Myoviridae | Unknown (not sequenced) | Dietes grandiflora soil | B. cenocepacia (K56-2), B. multivorans (C5274) [2 hosts] | Putatively obligately lytic; but not known conclusively due to challenges with sequencing | Not stable in haemolymph (3-log titer loss in 24 h). Likely uses LPS are a primary receptor [169,259]. | Reduces mortality in G. mellonella and completely clears K56-2 from haemolymph. Persists in murine lungs for at least three days after intraperitoneal or aerosolized delivery. NOID delivery produces significant (2.5-log) reductions in bacterial titer even 3 days after infection. Interacts synergistically with ciprofloxacin, meropenem and tetracycline based on plaque diameter and in vitro liquid growth reduction. Combinations of KS12 with minocycline and meropenem significantly reduce mortality in G. mellonella [169,170,244]. | |
| JG068 [259] | Podoviridae | Mguuvirus (Ralstonia phage ) | Sewage | B. multivorans (ATCC 17616), B. cenocepacia (K56-2, J2315, PC184), B. stabilis (LMG 14294), B. dolosa (AU0158, CEP021). [6 hosts] | Obligately lytic | LPS inner core is primary receptor [259]. | Reduces mortality in G. mellonella [259]. | |
| H111-1[261] | Myoviridae | Unclassified Myoviridae (Acidiothiobacillus phage AcaML1) | Lysogen of B. cenocepacia (H111) | B. multivorans (ATCC 17616, C5274), B. cenocepacia (C6433, 715J, K56-2, C1257, C5424, PC184, R161, R452, R750, R1284, R1285, R1314, R1434, R1619, R1882, R1883, R1884, R2314, S11528). [21 hosts] | Lysogeny-capable; integrates into arginine tRNA gene | LPS inner core is primary receptor [261]. | ||
| Young Lab | Bcep22 [256] | Podoviridae | Lessievirus (Burkholderia phage BcepMigl) | ? | B. cenocepacia (AU1054) [1 host] | Lysogeny-capable; integrates; lysogens are unstable (likely lost after one generation) | ||
| BcepIL02 [256] | Podoviridae | Lessievirus (Burkholderia phage DC1) | Corn rhizosphere | B. cenocepacia (PC184, AU1054) [2 hosts] | Lysogeny-capable; integrates; lysogens are unstable (likely lost after one generation) | Significantly reduces lung bacterial density in a murine infection model [168]. | ||
| BcepB1A [258] | Myoviridae | Unclassified Myoviridae (Burkholderia phage Bups1 | Unspecified soil | B. cenocepacia (s198B1A) [1 host] | Obligately lytic | |||
| Bcep43 [258] | Myoviridae | Naesvirus (Burkholderia phage Bcep781) | Unspecified soil | B. cenocepacia (74-34, Bcc43) [2 hosts] | Obligately lytic | |||
| Bcep1 [258] | Myoviridae | Naesvirus (Burkholderia phage BcepNY3) | Unspecified soil | B. cenocepacia (Bcc1) [1 host] | Obligately lytic | |||
| Bcep781 [258] | Myoviridae | Naesvirus (Burkholderia phage Bcep43) | Unspecified soil | B. cenocepacia (74-34, Bcc43) [2 hosts] | Obligately lytic | |||
| Govan Lab | NS1 [252] | Myoviridae | Unknown (not sequenced) | Lysogen of B. vietnamiensis (ATCC 29424) | B. cepacia (J2540, C2970), B. multivorans (C2775), B. cenocepacia (PC184, CEP511, C2836, C3166, LMG 18829, C3170), B. stabilis (C3171), B. vietnamiensis (ATCC 53266, ATCC 53267, C2973, LMG 16232, C2978), B. anthina (J2951, C1765), B. pyrrocinnia (C3918, C3930). [19 hosts] | Lysogeny-capable | LPS is likely a primary receptor [252]. | |
| NS2 [252] | Myoviridae | Unknown (not sequenced) | Lysogen of B. multivorans ATCC 17616 | B. cepacia (J2540, C2970), B. multivorans (C2775), B. cenocepacia (PC184, CEP511, ATCC17765, C1394, J415, BC7, K52-6, C6433, J2315, C2836, C3165, C3166, LMG 18829, C3170), B. vietnamiensis (ATCC 53266, ATCC 53267, C2973, LMG 16232, LMG 18836, ATCC 29424). [23 hosts] | Lysogeny-capable | LPS is likely a primary receptor [252]. | ||
| DK1 [255] | Siphoviridae | Unknown (not sequenced) | ? | B. multivorans (C2775), B. cenocepacia (J415, LMG 18829, ATCC 17765), B. vietnamiensis (C3177), B. pyrrocinnia (C3918) [6 hosts] | Lysogeny-capable | |||
| DK2/DK3 [255] | Myoviridae | Unknown (not sequenced) | Lysogen of B. cenocepacia (C3166) and B. stabilis (C3174) | B. cepacia (ATCC 17759), B. multivorans (C2775), B. cenocepacia (C1394, J2956, LMG 18829), B. stabilis (C3173), B. vietnamiensis (C3177) [7 hosts] | Lysogeny-capable | |||
| JB1 [255] | Myoviridae | Unknown (not sequenced) | Unspecified soil | B. cepacia (C2970), B. cenocepacia (J2956, LMG 18829, ATCC 17765), B. stabilis (LMG 18870), B. vietnamiensis (C3177), B. anthina (J2951, C1658, C1765), B. pyrrocinnia (C3909, C3918, C3930, C3993, C3995, C3997), B. ubonensis (E551). [16 hosts] | Unknown (not sequenced) | |||
| JB3 [255] | Siphoviridae | Unknown (not sequenced) | Unspecified plant rhizosphere | B. multivorans (C2775), B. cenocepacia (J2956, LMG 18829), B. anthina (C1658, C1765), B. ubonensis (E551) [6 hosts] | Unknown (not sequenced) | |||
| JB5 [255] | Myoviridae | Unknown (not sequenced) | Unspecified plant rhizosphere | B. cepacia (C2970), B. cenocepacia (J2956, C2836, LMG 18829, ATCC 17765), B. vietnamiensis (C3175, C3177), B. anthina (J2951, C1658, C1765), B. pyrrocinnia (C3909, C3918, C3830, C3993, C3995, C3997). [16 hosts] | Unknown (not sequenced) | |||
| RL1c [255] | Myoviridae | Unknown (not sequenced) | Unspecified plant rhizosphere | B. cepacia (C2970), B. multivorans (C2775), B. cenocepacia (J2956, ATCC 17765), B. stabilis (C3171), B. vietnamiensis (C2978, C3177), B. anthina (C1765). [8 hosts] | Unknown (not sequenced) | |||
| RL2 [255] | Myoviridae | Unknown (not sequenced) | Pond sediment | B. cenocepacia (J415, C1394, J2956, C2836, C3169, C3170), B. vietnamiensis (C3177), B. anthina (J2951, J2862), B. pyrrocinnia (C3918, C3930, C3993, C3995), B. ubonensis (E26). [15 hosts] | Unknown (not sequenced) | |||
| Others | CP75 [272] | Myoviridae | Unknown (not sequenced) | Lysogen of Pseudomonas cepacia (PCT1) | No known hosts | Lysogeny-capable | First identified phage of the Bcc [272]. | |
| BcP15 [273,274] | Siphoviridae | Unknown (not sequenced) | Lysogen of B. cepacia DR11 | No known hosts | Lysogeny-capable | |||
| AP3 [265] | Myoviridae | Kisquinquevirus (Burkholderia phage Mana) | Water sample | B. cenocepacia (5, 6, 10, 18, 20, 21, 7780, 1567, 1947, 39) [10 hosts] | Lysogeny-capable; integrates into tRNA genes | Reduces mortality in G. mellonella. [265] | ||
| G4P1 [275] | Myoviridae | Unclassified Myoviridae (Manheimia haemolytica phage 3927AP2) | Lysogen of B. vietnamiensis G4 | B. ambifaria (J82, R-8863, ATCC 51671, LMG-P 24640, LMG 17828, MVP/C1 64, MC40-6), B. cenocepacia (LMG 18829), B. contaminans (CEP0964), B. dolosa (AU0794, AU3556, AU1568, AU3960, AU4298, AU2130), B. vietnamiensis (LMG 18835, LMG 10929; CEP1224). [18 hosts] | Lysogeny-capable; integrates into arginine tRNA gene | |||
4.3. Experimental Bcc Phage Therapy In Vivo
5. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention Antibiotic Resistance Threats in the United States, 2019; AR Threats Report; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. The Review on Antimicrobial Resistance; Welcome Trust: London, UK, 2014. [Google Scholar]
- Council of Canadian Academies. When Antibiotics Fail. 2019. Available online: https://www.cca-reports.ca/reports/the-potential-socio-economic-impacts-of-antimicrobial-resistance-in-canada (accessed on 20 June 2021).
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. 10 × ′20 progress—Development of new drugs active against gram-negative bacilli: An update from the infectious diseases society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Brüssow, H.; Hendrix, R.W. Phage genomics. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Mahenthiralingam, E.; Urban, T.A.; Goldberg, J.B. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef]
- Burkholder, W. Sour skin, a bacterial rot of onion bulbs. Phytopathology 1950, 40, 115–117. [Google Scholar]
- Stanier, R.Y.; Palleroni, N.J.; Doudoroff, M. The aerobic pseudomonads: A taxonomic study. J. Gen. Microbiol. 1966, 43, 159–271. [Google Scholar] [CrossRef] [Green Version]
- Lessie, T.G.; Gaffney, T. Catabolic potential of pseudomonas cepacia. In The Biology of Pseudomonas; Sokatch, J.R., Ed.; Academic Press, Inc.: Orlando, FL, USA, 1986; pp. 439–481. [Google Scholar]
- Fiore, A.; Laevens, S.; Bevivino, A.; Dalmastri, C.; Tabacchioni, S.; Vandamme, P.; Chiarini, L. Burkholderia cepacia complex: Distribution of genomovars among isolates from the maize rhizosphere in Italy. Environ. Microbiol. 2001, 3, 137–143. [Google Scholar] [CrossRef]
- Miller, S.C.M.; LiPuma, J.J.; Parke, J.L. Culture-based and non-growth-dependent detection of the Burkholderia cepacia complex in soil environments. Appl. Environ. Microbiol. 2002, 68, 3750–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermis, K.; Vandamme, P.A.R.; Nelis, H.J. Burkholderia cepacia complex genomovars: Utilization of carbon sources, susceptibility to antimicrobial agents and growth on selective media. J. Appl. Microbiol. 2003, 95, 1191–1199. [Google Scholar] [CrossRef]
- Vonberg, R.P.; Gastmeier, P. Hospital-acquired infections related to contaminated substances. J. Hosp. Infect. 2007, 65, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, E.; Kosako, Y.; Oyaizu, H.; Yano, I.; Hotta, H.; Hashimoto, Y.; Ezaki, T.; Arakawa, M. Proposal of Burkholder gen. nov. and transfer of seven of the genus pseudomona homology group ii to the new genus, with type species Burkholderia cepecia. Microbiol. Immunol. 1992, 36, 1251–1275. [Google Scholar] [CrossRef]
- Gillis, M.; Van Van, T.; Bardin, R.; Goor, M.; Hebbar, P.; Willems, A.; Segers, P.; Kersters, K.; Heulin, T.; Fernandez, M.P. Polyphasic taxonomy in the genus Burkholderia leading to an emended description of the genus and proposition of Burkholderia vietnamiensis sp. nov. for N2-fixing isolates from rice in Vietnam. Int. J. Syst. Bacteriol. 1995, 45, 274–289. [Google Scholar] [CrossRef]
- Vandamme, P.; Holmes, B.; Vancanneyt, M.; Coenye, T.; Hoste, B.; Coopman, R.; Revets, H.; Lauwers, S.; Gillis, M.; Kersters, K.; et al. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int. J. Syst. Bacteriol. 1997, 47, 1188–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenye, T.; Mahenthiralingam, E.; Henry, D.; LiPuma, J.J.; Laevens, S.; Gillis, M.; Speert, D.P.; Vandamme, P. Burkholderia ambifaria sp. nov., a novel member of the Burkholderia cepacia complex including biocontrol and cystic fibrosis-related isolates. Int. J. Syst. Evol. Microbiol. 2001, 51, 1481–1490. [Google Scholar] [CrossRef] [Green Version]
- Coenye, T.; Vandamme, P.; Govan, J.R.W.; Lipuma, J.J. Taxonomy and identification of the Burkholderia cepacia complex. J. Clin. Microbiol. 2001, 39, 3427–3436. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, P.; Henry, D.; Coenye, T.; Nzula, S.; Vancanneyt, M.; LiPuma, J.J.; Speert, D.P.; Govan, J.R.W.; Mahenthiralingam, E. Burkholderia anthina sp. nov. and Burkholderia pyrrocinia, two additional Burkholderia cepacia complex bacteria, may confound results of new molecular diagnostic tools. FEMS Immunol. Med. Microbiol. 2002, 33, 143–149. [Google Scholar] [CrossRef] [Green Version]
- De Smet, B.; Mayo, M.; Peeters, C.; Zlosnik, J.E.A.; Spilker, T.; Hird, T.J.; Li Puma, J.J.; Kidd, T.J.; Kaestli, M.; Ginther, J.L.; et al. Burkholderia stagnalis sp. nov. and Burkholderia territorii sp. nov., two novel Burkholderia cepacia complex species from environmental and human sources. Int. J. Syst. Evol. Microbiol. 2015, 65, 2265–2271. [Google Scholar] [CrossRef]
- Depoorter, E.; Bull, M.J.; Peeters, C.; Coenye, T.; Vandamme, P.; Mahenthiralingam, E. Burkholderia: An update on taxonomy and biotechnological potential as antibiotic producers. Appl. Microbiol. Biotechnol. 2016, 100, 5215–5229. [Google Scholar] [CrossRef]
- Ong, K.S.; Aw, Y.K.; Lee, L.H.; Yule, C.M.; Cheow, Y.L.; Lee, S.M. Burkholderia paludis sp. nov., an antibiotic-siderophore producing novel Burkholderia cepacia complex species, isolated from malaysian tropical peat swamp soil. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, E.; Sant’Anna, F.H.; dos Passos, J.F.M.; Balsanelli, E.; de Baura, V.A.; de Oliveria Pedrosa, F.; de Souza, E.M.; Passaglia, L.M.P. Detection of misidentifications of species from the Burkholderia cepacia complex and description of a new member, the soil bacterium Burkholderia catarinensis sp. nov. Pathog. Dis. 2017, 75, 1–8. [Google Scholar] [CrossRef]
- Weber, C.F.; King, G.M. Volcanic soils as sources of novel CO-oxidizing Paraburkholderia and Burkholderia: Paraburkholderia hiiakae sp. nov., Paraburkholderia metrosideri sp. nov., Paraburkholderia paradisi sp. nov., Paraburkholderia peleae sp. nov., and Burkholderia alpina sp. nov. a Member of the Burkholderia cepacia complex. Front. Microbiol. 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, P.; Leguizamon, M.; Prieto, C.I.; Sousa, S.A.; Montanaro, P.; Draghi, W.O.; Stämmler, M.; Bettiol, M.; de Carvalho, C.C.C.R.; Palau, J.; et al. Burkholderia puraquae sp. nov., a novel species of the Burkholderia cepacia complex isolated from hospital settings and agricultural soils. Int. J. Syst. Evol. Microbiol. 2018, 68, 14–20. [Google Scholar] [CrossRef]
- Ballard, R.W.; Palleroni, N.J.; Doudoroff, M.; Stanier, R.Y. Taxonomy of the Aerobic Pseudomonads: Pseudomonas cepacia, P. marginata, P. allicola, and P. caryophylli. J. Gen. Microbiol. 1970, 60, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Furuya, N.; Iiyama, K.; Ueda, Y.; Matsuyama, N. Reaction of tobacco and rice leaf tissue infiltrated with Burkholderia glumae or B. gladioli. J. Fac. Agric. Kyushu Univ. 1997, 42, 43–51. [Google Scholar] [CrossRef]
- Goto, K.; Ohanta, K. New bacterial diseases of rice—bacterial brown stripe and bacterial grain rot. Ann. Phytopathol. Soc. Jpn. 1956, 21, 46–47. [Google Scholar]
- Azegami, K.; Nishiyama, K.; Watanabe, Y.; Kadota, I.; Ohuch, A.; Fukazawa, C. Pseudomonas plantarii sp. nov., the causal agent of rice seedling blight. Int. J. Syst. Bacteriol. 1987, 37, 475. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, R.; Shahjahan, A.K.M.; Yuan, X.L.; Dickstein, E.R.; Groth, D.E.; Clark, C.A.; Cartwright, R.D.; Rush, M.C. Burkholderia glumae and B. gladioli cause bacterial panicle blight in rice in the Southern United States. Plant. Dis. 2009, 93, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, L.; Bevivino, A.; Tabacchioni, S.; Dalmastri, C. Inoculation of Burkholderia cepacia, Pseudomonas fluorescens and Enterobacter sp. on Sorghum bicolor: Root colonization and plant growth promotion of dual strain inocula. Soil Biol. Biochem. 1998, 30, 81–87. [Google Scholar] [CrossRef]
- Reis, V.M.; Baldani, J.I.; Baldani, V.L.D.; Dobereiner, J. Biological dinitrogen fixation in gramineae and palm trees. CRC. Crit. Rev. Plant. Sci. 2000, 19, 227–247. [Google Scholar] [CrossRef]
- Perin, L.; Martínez-Aguilar, L.; Castro-González, R.; Estrada-De Los Santos, P.; Cabellos-Avelar, T.; Guedes, H.V.; Reis, V.M.; Caballero-Mellado, J. Diazotrophic Burkholderia species associated with field-grown maize and sugarcane. Appl. Environ. Microbiol. 2006, 72, 3103–3110. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.H.; Chen, F.F.; Wang, C.E.; Fu, H.H.; Fang, X.Q.; Ye, J.R.; Shi, J.Y. Indole-3-acetic acid in Burkholderia pyrrocinia JK-SH007: Enzymatic identification of the indole-3-acetamide synthesis pathway. Front. Microbiol. 2019, 10, 2559. [Google Scholar] [CrossRef] [Green Version]
- Thomashow, L.; Weller, D. Application of fluorescent pseudomonads to control root diseases of wheat and some mechanisms of disease suppression. In Proceedings of the Biological Control of Soil-Borne Plant Pathogens; Hornby, D., Ed.; CABI International: Wallingford, UK, 1990; pp. 109–122. [Google Scholar]
- Haas, D.; Défago, G. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 2005, 3, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Janisiewicz, W.J.; Roitman, J. Biological control of blue mold and gray mold on apple and pear with Pseudomonas cepacia. Phytopathology 1988, 78, 1697–1700. [Google Scholar] [CrossRef]
- Hebbar, K.; Atkinson, D.; Tucker, W.; Dart, P.J. Suppression of Fusarium moniliforme by maize root-associated Pseudomonas cepacia. Soil Biol. Biochem. 1992, 24, 1009–1020. [Google Scholar] [CrossRef]
- Chang, H.K.; Zylstra, G.J. Role of quinolinate phosphoribosyl transferase in degradation of phthalate by Burkholderia cepacia DBO. J. Bacteriol. 1999, 181, 3069–3075. [Google Scholar] [CrossRef] [Green Version]
- Bick, J.A.; Dennis, J.J.; Zylstra, G.J.; Nowack, J.; Leustek, T. Identification of a new class of 5′-adenylylsulfate (APS) reductases from sulfate-assimilating bacteria. J. Bacteriol. 2000, 182, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangodkar, U.; Chapman, P.; Chakrabarty, A. Cloning, physical mapping and expression of chromosomal genes specifying degradation of the herbicide 2,4,5-T by Pseudomonas cepacia AC. Gene 1988, 71, 267–277. [Google Scholar] [CrossRef]
- Yee, D.C.; Maynard, J.A.; Wood, T.K. Rhizoremediation of trichloroethylene by a recombinant, root-colonizing Pseudomonas fluorescens strain expressing toluene ortho-monooxygenase constitutively. Appl. Environ. Microbiol. 1998, 64, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Okoh, A.; Ajisebutu, S.; Babalola, G.; Trejo-Hernandcz, M.R. Potential of Burkholderia cepacia RQ1 in the biodegradation of heavy crude oil. Int. Microbiol. 2001, 4, 83–87. [Google Scholar] [CrossRef]
- Zylstra, G.J.; Olsen, R.H.; Ballou, D.P. Genetic organization and sequence of the Pseudomonas cepacia genes for the alpha and beta subunits of protocatechuate 3,4-dioxygenase. J. Bacteriol. 1989, 171, 5915–5921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerben, J.; Olsen, R.; Ballou, D. Cloning, expression, and regulation of the Pseudomonas cepacia Protocatechuate 3,4-Dioxygenase Genes. J. Bacteriol. 1989, 171, 5907–5914. [Google Scholar]
- Chang, H.K.; Mohseni, P.; Zylstra, G.J. Characterization and regulation of the genes for a novel anthranilate 1,2-dioxygenase from Burkholderia cepacia DBO1. J. Bacteriol. 2003, 185, 5871–5881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.K.; Dennis, J.J.; Zylstra, G.J. Involvement of two transport systems and a specific porin in the uptake of phthalate by Burkholderia spp. J. Bacteriol. 2009, 191, 4671–4673. [Google Scholar] [CrossRef] [Green Version]
- Parke, J.L.; Gurian-Sherman, D. Diversity of the Burkholderia Cepacia complex and implications for risk assessment of biological control strains. Annu. Rev. Phytopathol. 2001, 39, 225–258. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Baldwin, A.; Dowson, C.G. Burkholderia cepacia complex bacteria: Opportunistic pathogens with important natural biology. J. Appl. Microbiol. 2008, 104, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Isles, A.; Maclusky, I.; Corey, M.; Gold, R.; Prober, C.; Fleming, P.; Levison, H. Pseudomonas cepacia infection in cystic fibrosis: An emerging problem. J. Pediatr. 1984, 104, 206–210. [Google Scholar] [CrossRef]
- O’Neil, K.M.; Herman, J.H.; Modlin, J.F.; Moxon, E.R.; Winkelstein, J.A. Pseudomonas cepacia: An emerging pathogen in chronic granulomatous disease. J. Pediatr. 1986, 108, 940–942. [Google Scholar] [CrossRef]
- Winkelstein, J.; Marino, M.; Johnston, R.; Boyle, J.; Curnutte, J.; Gallin, J.; Malech, H.; Holland, S.; Ochs, H.; Quie, P.; et al. Chronic Granulomatous Disease—Report on a National Registry of 368 Patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef]
- Bylund, J.; Campsall, P.A.; Ma, R.C.; Conway, B.-A.D.; Speert, D.P. Burkholderia cenocepacia Induces neutrophil necrosis in chronic granulomatous disease. J. Immunol. 2005, 174, 3562–3569. [Google Scholar] [CrossRef] [Green Version]
- Tablan, O.; Chorba, T.; Schidlow, D.; White, J.; Hardy, K.; Gilligan, P.; Meade Morgan, W.; Carson, L.; Martone, W.; Jason, J.; et al. Pseudomonas cepacia colonization in patients with cystic fibrosis: Risk factors and clinical outcome. J. Pediatr. 1985, 107, 382–387. [Google Scholar] [CrossRef]
- Liou, T.; Adler, F.; FitzSimmons, S.; Cahill, B.; Hibbs, J.; Marshall, B. Predictive 5-Year Survivorship Model of Cystic Fibrosis. Am. J. Epidemiol. 2001, 153, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.H.; Stothard, P.; Dennis, J.J. Genomic analysis and relatedness of P2-like phages of the Burkholderia cepacia complex. BMC Genom. 2010, 11, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.M.; Dodd, M.E.; Govan, J.R.W.; Barcus, V.; Doherty, C.J.; Morris, J.; Webb, A.K. Burkholderia cenocepacia and Burkholderia multivorans: Influence on survival in cystic fibrosis. Thorax 2004, 59, 948–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govan, J.; Doherty, C.J.; Nelson, J.; Brown, P.; Greening, A.; Maddison, J.; Dodd, M.; Webb, A. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993, 342, 15–19. [Google Scholar] [CrossRef]
- Coenye, T.; LiPuma, J.J. Population structure analysis of Burkholderia cepacia genomovar III: Varying degrees of genetic recombination characterize major clonal complexes. Microbiology 2003, 149, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Coenye, T.; Spilker, T.; Van Schoor, A.; LiPuma, J.J.; Vandamme, P. Recovery of Burkholderia cenocepacia strain PHDC from cystic fibrosis patients in Europe. Thorax 2004, 59, 952–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duff, A. Psychological consequences of segregation resultin from chronic Burkholderia cepacia infection in adults with CF. Thorax 2002, 57, 756–758. [Google Scholar] [CrossRef] [Green Version]
- Saiman, L.; Siegel, J. Infection control in cystic fibrosis. Clin. Microbiol. Rev. 2004, 17, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Garber, E.; Saiman, L. Survey of infection control policies for patients with cystic fibrosis in the United States. Am. J. Infect. Control. 2008, 36, 220–222. [Google Scholar] [CrossRef]
- Drabick, J.; Gracely, E.; Heidecker, G.; LiPuma, J. Survival of Burkholderia cepacia on environmental surfaces. J. Hosp. Infect. 1996, 32, 267–276. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, J.M.; Kweon, O.; Kim, S.J.; Jones, R.C.; Woodling, K.; Gamboa da Costa, G.; LiPuma, J.J.; Hussong, D.; Marasa, B.S.; et al. Intrinsic resistance of Burkholderia cepacia complex to benzalkonium chloride. mBio 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LiPuma, J.J.; Spilker, T.; Coenye, T.; Gonzalez, C.F. An epidemic Burkholderia cepacia complex strain identified in soil. Lancet 2002, 359, 2002–2003. [Google Scholar] [CrossRef]
- LiPuma, J.J.; Spilker, T.; Gill, L.H.; Campbell, P.W.; Liu, L.; Mahenthiralingam, E. Disproportionate distribution of Burkholderia cepacia complex species and transmissibility markers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2001, 164, 92–96. [Google Scholar] [CrossRef]
- Reik, R.; Spilker, T.; LiPuma, J.J. Distribution of Burkholderia cepacia complex species among isolates recovered from persons with or without cystic fibrosis. J. Clin. Microbiol. 2005, 43, 2926–2928. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Hoffman, L.R.; Whiteley, M.; Daniels, T.W.V.; Carroll, M.P.; Bruce, K.D. Revealing the dynamics of polymicrobial infections: Implications for antibiotic therapy. Trends Microbiol. 2010, 18, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragonzi, A.; Farulla, I.; Paroni, M.; Twomey, K.B.; Pirone, L.; Lorè, N.I.; Bianconi, I.; Dalmastri, C.; Ryan, R.P.; Bevivino, A. Modelling co-infection of the cystic fibrosis lung by Pseudomonas aeruginosa and Burkholderia cenocepacia reveals influences on biofilm formation and host response. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Spilker, T.; LiPuma, J.J. Simultaneous coinfection by multiple strains during Burkholderia cepacia complex infection in cystic fibrosis. Diagn. Microbiol. Infect. Dis. 2006, 54, 95–98. [Google Scholar] [CrossRef]
- Greenberg, D.E.; Goldberg, J.B.; Stock, F.; Murray, P.R.; Holland, S.M.; LiPuma, J.J. Recurrent Burkholderia infection in patients with chronic granulomatous disease: 11-year experience at a large referral center. Clin. Infect. Dis. 2009, 48, 1577–1579. [Google Scholar] [CrossRef] [Green Version]
- Torbeck, L.; Raccasi, D.; Guilfoyle, D.E.; Friedman, R.L.; Hussong, D. Burkholderia cepacia: This decision is overdue. PDA J. Pharm. Sci. Technol. 2011, 65, 535–543. [Google Scholar] [CrossRef] [Green Version]
- El Chakhtoura, N.G.; Saade, E.; Wilson, B.M.; Perez, F.; Papp-Wallace, K.M.; Bonomo, R.A. A 17-year nationwide study of Burkholderia cepacia complex bloodstream infections among patients in the United States Veterans Health Administration. Clin. Infect. Dis. 2017, 65, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Bye, P.T.B. Mucociliary clearance in cystic fibrosis. Pediatr. Pulmonol. 2002, 33, 293–306. [Google Scholar] [CrossRef]
- Verkman, A.S.; Song, Y.; Thiagarajah, J.R. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am. J. Physiol. Physiol. 2003, 284, C2–C15. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respir. J. 2017, 49, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Govan, J.R.W.; Deretic, V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 1996, 60, 539–574. [Google Scholar] [CrossRef]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.; Freedman, S. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- LiPuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, T.; Gifford, A.H.; Sabadosa, K.A.; Quinton, H.B.; Knapp, E.A.; Goss, C.H.; Marshall, B.C. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: Survival analysis of the cystic fibrosis foundation patient registry. Ann. Intern. Med. 2014, 161, 233–241. [Google Scholar] [CrossRef]
- Johnston, R.; Newman, S. Chronic granulomatous disease. Pediatr. Clin. North Am. 1977, 24, 365–376. [Google Scholar] [CrossRef]
- Van den Berg, J.M.; van Koppen, E.; Åhlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Espanñol, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic granulomatous disease: The European experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef]
- Gallin, J.; Malech, H.; Weening, R.; Curnutte, J.; Quie, P.; Jaffe, H.; Ezekowitz, R. A Controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N. Engl. J. Med. 1991, 324, 509–516. [Google Scholar]
- Cale, C.M.; Jones, A.M.; Goldblatt, D. Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clin. Exp. Immunol. 2000, 120, 351–355. [Google Scholar] [CrossRef]
- Pao, M.; Wiggs, E.; Anastacio, M.; Hyun, J.; DeCarlo, E.; Miller, J.; Anderson, V.; Malech, H.; Gallin, J.; Holland, S. Cognitive function in patients with chronic granulomatous disease: A preliminary report. Psychosomatics 2004, 45, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LiPuma, J.J. Burkholderia cepacia: Management issues and new insights. Clin. Chest Med. 1998, 19, 473–486. [Google Scholar] [CrossRef]
- Aaron, S.D.; Ferris, W.; Henry, D.A.; Speert, D.P.; Macdonald, N.E. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am. J. Respir. Crit. Care Med. 2000, 162, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Y.; Tabibi, S.; Alba, L.; Garber, E.; Saiman, L. Antimicrobial susceptibility and synergy studies of Burkholderia cepacia complex isolated from patients with cystic fibrosis. Antimicrob. Agents Chemother. 2007, 51, 1085–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loutet, S.A.; Valvano, M.A. Extreme antimicrobial peptide and polymyxin B resistance in the genus Burkholderia. Front. Cell. Infect. Microbiol. 2011, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Buroni, S.; Pasca, M.R.; Flannagan, R.S.; Bazzini, S.; Milano, A.; Bertani, I.; Venturi, V.; Valvano, M.A.; Riccardi, G. Assessment of three resistance-nodulation-cell division drug efflux transporters of Burkholderia cenocepacia in intrinsic antibiotic resistance. BMC Microbiol. 2009, 9, 1–11. [Google Scholar] [CrossRef]
- Chiesa, C.; Labrozzi, P.H.; Aronoff, S.C. Decreased baseline β-Lactamase production and inducibility associated with increased piperacillin susceptibility of Pseudomonas cepacia isolated from children with cystic fibrosis. Pediatr. Res. 1986, 20, 1174–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everaert, A.; Coenye, T. Effect of β-Lactamase inhibitors on in vitro activity of β-Lactam antibiotics against Burkholderia cepacia complex species. Antimicrob. Resist. Infect. Control 2016, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Beckman, W.; Lessie, T.G. Response of Pseudomonas cepacia to β-lactam antibiotics: Utilization of penicillin G as the carbon source. J. Bacteriol. 1979, 140, 1126–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.L.; Hedin, L.A.; Lien, D.M. Chloramphenicol resistance in Pseudomonas cepacia because of decreased permeability. Antimicrob. Agents Chemother. 1989, 33, 136–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinion-Dubiel, A.D.; Goldberg, J.B. Lipopolysaccharide of Burkholderia cepacia complex. J. Endotoxin Res. 2003, 9, 201–213. [Google Scholar] [CrossRef]
- Ortega, X.P.; Cardona, S.T.; Brown, A.R.; Loutet, S.A.; Flannagan, R.S.; Campopiano, D.J.; Govan, J.R.W.; Valvano, M.A. A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J. Bacteriol. 2007, 189, 3639–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.L.; Wadsworth, C.D.; Barry, J.J.; Goodall, C.P. Nucleotide sequence analysis of a gene from Burkholderia (Pseudomonas) cepacia encoding an outer membrane lipoprotein involved in multiple antibiotic resistance. Antimicrob. Agents Chemother. 1996, 40, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, X.Z.; Poole, K. Fluoroquinolone susceptibilities of efflux-mediated multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia. J. Antimicrob. Chemother. 2001, 48, 549–552. [Google Scholar] [CrossRef]
- Podnecky, N.L.; Rhodes, K.A.; Schweizer, H.P. Efflux pump-mediated drug resistance in burkholderia. Front. Microbiol. 2015, 6, 305. [Google Scholar] [CrossRef] [Green Version]
- Holden, M.T.G.; Seth-Smith, H.M.B.; Crossman, L.C.; Sebaihia, M.; Bentley, S.D.; Cerdeño-Tárraga, A.M.; Thomson, N.R.; Bason, N.; Quail, M.A.; Sharp, S.; et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 2009, 91, 261–277. [Google Scholar] [CrossRef] [Green Version]
- Bazzini, S.; Udine, C.; Sass, A.; Pasca, M.R.; Longo, F.; Emiliani, G.; Fondi, M.; Perrin, E.; Decorosi, F.; Viti, C.; et al. Deciphering the role of RND efflux transporters in burkholderia cenocepacia. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Nair, B.M.; Cheung, K.J.; Griffith, A.; Burns, J.L. Salicylate induces an antibiotic efflux pump in burkholderia cepacia complex genomovar III (B. cenocepacia). J. Clin. Invest. 2004, 113, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Tseng, S.P.; Tsai, W.C.; Liang, C.Y.; Lin, Y.S.; Huang, J.W.; Chang, C.Y.; Tyan, Y.C.; Lu, P.L. The contribution of antibiotic resistance mechanisms in clinical Burkholderia cepacia complex isolates: An emphasis on efflux pump activity. PLoS ONE 2014, 9, e104986. [Google Scholar] [CrossRef]
- Speert, D.P. Advances in Burkholderia cepacia complex. Paediatr. Respir. Rev. 2002, 3, 230–235. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef]
- Blumer, J.L.; Saiman, L.; Konstan, M.W.; Melnick, D. The efficacy and safety of meropenem and tobramycin vs ceftazidime and tobramycin in the treatment of acute pulmonary exacerbations in patients with cystic fibrosis. Chest 2005, 128, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Peeters, E.; Nelis, H.J.; Coenye, T. In vitro activity of ceftazidime, ciprofloxacin, meropenem, minocycline, tobramycin and trimethoprim/sulfamethoxazole against planktonic and sessile Burkholderia cepacia complex bacteria. J. Antimicrob. Chemother. 2009, 64, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankin, E. L’action bactericide des eaux de la Jumna et du Gange sur le vibrion du cholera. Ann. Inst. Pasteur 1896, 10, 511–523. [Google Scholar]
- Gamaleya, N. Bacteriolysins-ferments destroying bacteria. Russ. Arch. Pathol. Clin. Med. Bacteriol. 1898, 6, 607–613. [Google Scholar]
- Twort, F.W. An Investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- Summers, W.C. Bacteriophage therapy. Annu. Rev. Microbiol. 2001, 55, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N. Phage Therapy: History from Twort and d’Herelle Through Soviet Experience to Current Approaches, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 83, pp. 3–40. [Google Scholar] [CrossRef]
- D’Herelle, F. Sur un microbe invisible antagoniste des bacilles dysenteriques. Comptes Rendus Acad. Sci. 1917, 165, 373–375. [Google Scholar]
- D’Herelle, F.; Eliava, G. Unicite du bacteriae sur la lysine du bacteriophage. Comptes Rendus Soc. Biol. 1921, 84, 701–702. [Google Scholar]
- D’Herelle, F.; Rakieten, M.L. The Adaptation of a Staphylococcus Bacteriophage to an artificially produced anti-bacteriophagic serum. J. Immunol. 1935, 28, 413–423. [Google Scholar]
- Bruynoghe, R.; Maisin, J. Essais de therapeutique au moyen du bacteriophage. Comptes Rendus Soc. Biol. 1921, 85, 1120–1121. [Google Scholar]
- Schless, R. Staphylococcus Aureus Meningitis: Treatment with specific bacteriophage. Am. J. Dis. Child. 1932, 44, 813–822. [Google Scholar] [CrossRef]
- Straub, M.; Applebaum, M. Studies on commercial bacteriophage products. JAMA J. Am. Med. Assoc. 1933, 100, 110–113. [Google Scholar] [CrossRef]
- Merril, C.R.; Scholl, D.; Adhya, S.L. The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2003, 2, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, M.D.; Bayne-Jones, S. Bacteriophage therapy: Review of the principles and results of the use of bacteriophages in the treatment of infections. JAMA J. Am. Med. Assoc. 1934, 23, 1769–1939. [Google Scholar] [CrossRef]
- Krueger, A.P.; Scribner, J.E. The Bacteriophage: Its nature and its therapeutic use. JAMA J. Am. Med. Assoc. 1941, 19, 2269–2277. [Google Scholar] [CrossRef]
- Rakieten, M.L. Studies with Staphyloccocus bacteriophage I. The preparation of polyvalent staphylococcus bacteriophage. Yale, J. Biol. Med. 1932, 4, 807–818. [Google Scholar]
- Sapir, I. Observations and recommendations related to phage therapy of dysentery. In Proceedings of Moscow Institute of Infectious Diseases after II Mechnikov; 1939; pp. 135–151. [Google Scholar]
- Vlasov, K.; Artemenko, E. Treatment of chronic dysentery. Sov. Med. 1946, 10, 22–28. [Google Scholar]
- Krestovnikova, V. Phage treatment and phage prophylactics and their approval in the works of the Soviet researchers. J. Microbiol. Epidemiol. Immunol. 1947, 3, 56–65. [Google Scholar]
- Duplessis, C.; Biswas, B.; Hanisch, B.; Perkins, M.; Henry, M.; Quinones, J.; Wolfe, D.; Estrella, L.; Hamilton, T. Refractory Pseudomonas bacteremia in a 2-year-old sterilized by bacteriophage therapy. J. Pediatric Infect. Dis. Soc. 2018, 7, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.L.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M.; et al. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am. J. Transplant. 2019, 19, 2631–2639. [Google Scholar] [CrossRef]
- Babalova, E.; Katsitadze, K.; Sakvarelidze, L.; Imnaishvili, N.; Sharashidze, T.; Badashvili, V.; Kiknadze, G.; Meipariani, A.; Gendzekhadze, N.; Machavariani, E.; et al. Preventive value of dried dysentery bacteriophage. Zhurnal Mikrobiol. Epidemiol. I Immunol. 1968, 45, 143–145. [Google Scholar]
- Sayamov, R. Treatment and prophylaxis of cholera with bacteriophage. Bull. World Health Organ. 1963, 28, 361–367. [Google Scholar]
- Butler, M.S.; Blaskovich, M.A.; Cooper, M.A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 2013, 66, 571–591. [Google Scholar] [CrossRef] [Green Version]
- Slopek, S.; Weber-Dabrowska, B.; Dabrowski, M.; Kucharewicz-Krukowska, A. Results of bacteriophage treatment of suppurative bacterial infections in the years 1981–1986. Arch. Immunol. Ther. Exp. 1986, 35, 569–583. [Google Scholar]
- Soothill, J.S. Treatment of experimental infections of mice with bacteriophages. J. Med. Microbiol. 1992, 37, 258–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soothill, J.S. Bacteriophage prevents destruction of skin grafts by Pseudomonas aeruginosa. Burns 1994, 20, 209–211. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B. Successful treatment of experimental Escherichia coli infections in mice using phage: Its general superiority over antibiotics. J. Gen. Microbiol. 1982, 128, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Williams Smith, H.; Huggins, M.B. Effectiveness of phages in treating experimental Escherichia coli diarhoea in calves, piglets and lambs. J. Gen. Microbiol. 1983, 129, 2659–2675. [Google Scholar] [CrossRef] [Green Version]
- Williams Smith, H.; Huggins, M.B.; Shaw, K.M. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophages. J. Gen. Microbiol. 1987, 133, 1111–1126. [Google Scholar] [CrossRef] [Green Version]
- Bogovadzova, G.; Voroshilova, N.; Bondarenko, V. The efficacy of Klebsiella pneumoniae bacteriophage in the therapy of experimental Klebsiella infection. Zhurnal Mikrobiol. Epidemiol. I Immunol. 1991, 4, 5–8. [Google Scholar]
- Bogovadzova, G.; Voroshilova, N.; Bondarenko, V.; Gorbatkova, G.; Afanaseva, E.; Kazakova, T. Immunobiological properties and therapeutic effectiveness of preparations from Klebsiella bacteriophages. Zhurnal Mikrobiol. Epidemiol. I Immunol. 1992, 3, 30–33. [Google Scholar]
- Wilhelm, S.; Suttle, C. Viruses and nutrient cycles in the sea. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, R.W. Bacteriophages: Evolution of the majority. Theor. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
- Proctor, L.M.; Fuhrman, J.A. Viral mortality in bacteria and cyanobacteria. Nature 1990, 343, 60–62. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Calendar, R. The Bacteriophages, 2nd ed.; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Ackermann, H.W. Bacteriophage observations and evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Campbell, A. The future of bacteriophage biology. Nat. Rev. Genet. 2003, 4, 471–477. [Google Scholar] [CrossRef]
- Hatfull, G.; Hendrix, R. Bacteriophages and their Genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, A.A. Bacteriophage Receptors. Annu. Rev. Microbiol. 1973, 27, 205–241. [Google Scholar] [CrossRef]
- Adam, G.; Delbruck, M. Reduction of dimensionality in biological diffusion processes. Struct. Chem. Mol. Biol. 1968, 198, 198–215. [Google Scholar]
- Casjens, S.R.; Molineux, I.J. Short noncontractile tail machines: Adsorption and DNA delivery by podoviruses. Adv. Exp. Med. Biol. 2012, 726, 143–179. [Google Scholar]
- Rakhuba, D.; Kolomiets, E.; Szwajcer Dey, E.; Novik, G. Bacteriophage Receptors, Mechanisms of Phage Adsorption and Penetration into Host Cell. Pol. J. Microbiol. 2010, 59, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; Mesyanzhinov, V.V.; Arisaka, F.; Leiman, P.G. The bacteriophage T4 DNA injection machine. Curr. Opin. Struct. Biol. 2004, 14, 171–180. [Google Scholar] [CrossRef]
- Sharma, S.; Chatterjee, S.; Datta, S.; Prasad, R.; Dubey, D.; Prasad, R.K.; Vairale, M.G. Bacteriophages and its applications: An overview. Folia Microbiol. 2017, 62, 17–55. [Google Scholar] [CrossRef]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Evolution of superinfection immunity in cluster A mycobacteriophages. mBio 2019, 10, e00971-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronayne, E.A.; Wan, Y.C.S.; Boudreau, B.A.; Landick, R.; Cox, M.M. P1 Ref Endonuclease: A Molecular Mechanism for Phage-Enhanced Antibiotic Lethality. PLoS Genet. 2016, 12, e1005797. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M.; Strauch, E. Phage therapy: Facts and fiction. Int. J. Med. Microbiol. 2006, 296, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.H.; Seed, K.D.; Stothard, P.; Dennis, J.J. Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J. Virol. 2010, 84, 1276–1288. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Carmody, L.A.; Gill, J.J.; Summer, E.J.; Sajjan, U.S.; Gonzalez, C.F.; Young, R.F.; Lipuma, J.J. Efficacy of bacteriophage therapy in a model of burkholderia cenocepacia pulmonary infection. J. Infect. Dis. 2010, 201, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seed, K.D.; Dennis, J.J. Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob. Agents Chemother. 2009, 53, 2205–2208. [Google Scholar] [CrossRef] [Green Version]
- Semler, D.D.; Goudie, A.D.; Finlay, W.H.; Dennis, J.J. Aerosol phage therapy efficacy in Burkholderia cepacia complex respiratory infections. Antimicrob. Agents Chemother. 2014, 58, 4005–4013. [Google Scholar] [CrossRef] [Green Version]
- James, C.E.; Davies, E.V.; Fothergill, J.L.; Walshaw, M.J.; Beale, C.M.; Brockhurst, M.A.; Winstanley, C. Lytic activity by temperate phages of Pseudomonas aeruginosa in long-term cystic fibrosis chronic lung infections. ISME J. 2015, 9, 1391–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, J.G.; Peters, D.L.; Dennis, J.J. Identification and characterization of type IV Pili as the cellular receptor of broad host range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses 2018, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Der Torossian Torres, M.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance Through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469. [Google Scholar] [CrossRef]
- Seed, K.D.; Dennis, J.J. Isolation and characterization of bacteriophages of the Burkholderia cepacia complex. FEMS Microbiol. Lett. 2005, 251, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.J.; Kenna, D.; Bartholdson, J.; Campopiano, D.J.; Govan, J.R.W. Temperate bacteriophages DK4 and BcepMu from Burkholderia cenocepacia J2315 are identical. FEMS Immunol. Med. Microbiol. 2005, 45, 349–350. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; Lynch, K.H.; Stothard, P.; Dennis, J.J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. BioEssays 2015, 37, 6–9. [Google Scholar] [CrossRef]
- Carlton, R.M. Phage therapy: Past history and future prospects. Arch. Immunol. Ther. Exp. 1999, 47, 267–274. [Google Scholar]
- Skurnik, M.; Pajunen, M.; Kiljunen, S. Biotechnological challenges of phage therapy. Biotechnol. Lett. 2007, 29, 995–1003. [Google Scholar] [CrossRef]
- Rhoads, D.; Wolcott, R.; Kuskowski, M.; Wolcott, B.; Ward, L.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Prasad, Y. Efficacy of polyvalent bacteriophage P-27/HP to control multidrug resistant staphylococcus aureus associated with human infections. Curr. Microbiol. 2011, 62, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Thimmapuram, J.; Zhang, J.; Collings, C.K.; Bhide, K.; Schmidt, K.; Ebner, P.D. The impact of orally administered phages on host immune response and surrounding microbial communities. Bacteriophage 2016, 6, e1211066. [Google Scholar] [CrossRef] [Green Version]
- Cieplak, T.; Soffer, N.; Sulakvelidze, A.; Nielsen, D.S. A bacteriophage cocktail targeting Escherichia coli reduces E. coli in simulated gut conditions, while preserving a non-targeted representative commensal normal microbiota. Gut Microbes 2018, 9, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [Green Version]
- McCallin, S.; Alam Sarker, S.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From MetaGenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.A.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: A randomized trial in children from Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Slama, T.G.; Amin, A.; Brunton, S.A.; File, T.M.; Milkovich, G.; Rodvold, K.A.; Sahm, D.F.; Varon, J.; Weiland, D. A clinician’s guide to the appropriate and accurate use of antibiotics: The Council for Appropriate and Rational Antibiotic Therapy (CARAT) criteria. Am. J. Med. 2005, 118, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S. Phage Therapy in Clinical Practice: Treatment of Human Infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent diabetic toe ulcers: A case series. J. Wound Care 2016, 25, S27–S33. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Änggård, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; A preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kvachadze, L.; Balarjishvili, N.; Meskhi, T.; Tevdoradze, E.; Skhirtladze, N.; Pataridze, T.; Adamia, R.; Topuria, T.; Kutter, E.; Rohde, C.; et al. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb. Biotechnol. 2011, 4, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage Therapy for limb-threatening prosthetic knee klebsiella pneumoniae infection: Case report and in vitro characterization of anti-biofilm activity. Clin. Infect. Dis. 2020, 1–8. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef]
- Atterbury, R.J.; Van Bergen, M.A.P.; Ortiz, F.; Lovell, M.A.; Harris, J.A.; De Boer, A.; Wagenaar, J.A.; Allen, V.M.; Barrow, P.A. Bacteriophage therapy to reduce Salmonella colonization of broiler chickens. Appl. Environ. Microbiol. 2007, 73, 4543–4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef]
- Azam, A.H.; Tanji, Y. Bacteriophage-host arm race: An update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J.; et al. Personalized therapeutic cocktail of wild environmental phages rescues mice from acinetobacter baumannii wound infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raya, R.; H’bert, E. Isolation of Phage via Induction of Lysogens. In Bacteriophages. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 23–32. [Google Scholar]
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.; Wong, S.; St. Jean, J.; Slavcev, R. Bacteriophage interactions with mammalian tissue: Therapeutic applications. Adv. Drug Deliv. Rev. 2019, 145, 4–17. [Google Scholar] [CrossRef]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. BioEssays 2011, 33, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semler, D.D.; Lynch, K.H.; Dennis, J.J. The promise of bacteriophage therapy for Burkholderia cepacia complex respiratory infections. Front. Cell. Infect. Microbiol. 2011, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary Rationale for Phages as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Olszak, T.; Arabski, M.; Wasik, S.; Majkowska-Skrobek, G.; Augustyniak, D.; Gula, G.; Briers, Y.; Jang, H.B.; Vandenheuvel, D.; et al. Characterization of the newly isolated lytic bacteriophages KTN6 and KT28 and their efficacy against pseudomonas aeruginosa biofilm. PLoS ONE 2015, 10, e0127603. [Google Scholar] [CrossRef]
- Pires, D.P.; Oliveira, H.; Melo, L.D.R.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [Green Version]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a broad-range bacteriophage cocktail that reduces pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef]
- Kurzȩpa, A.; Dąbrowska, K.; Skaradziński, G.; Górski, A. Bacteriophage interactions with phagocytes and their potential significance in experimental therapy. Clin. Exp. Med. 2009, 9, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Nafissi, N.; Wasfi, O.; Sheldon, K.; Wettig, S.; Slavcev, R. Immunocompatibility of bacteriophages as nanomedicines. J. Nanotechnol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Miernikiewicz, P.; Dabrowska, K.; Piotrowicz, A.; Owczarek, B.; Wojas-Turek, J.; Kicielińska, J.; Rossowska, J.; Pajtasz-Piasecka, E.; Hodyra, K.; Macegoniuk, K.; et al. T4 Phage and Its Head Surface Proteins Do Not Stimulate Inflammatory Mediator Production. PLoS ONE 2013, 8, e71036. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Davis, S.D.; Wedgwood, R.J. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J. Clin. Invest. 1971, 50, 2559–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.S.; Kaido, T.; Carrier, E. Immunological factors that affect the in vivo fate of T7 phage in the mouse. J. Virol. Methods 2004, 115, 99–104. [Google Scholar] [CrossRef]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jonczyk-Matysiak, E.; Lecion, D.; Kazmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian Host-Versus-Phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharewicz-Krukowska, A.; Slopek, S. Immunogenic effect of bacteriophage in patients subjected to phage therapy. Arch. Immunol. Ther. Exp. 1987, 35, 553–561. [Google Scholar]
- Lusiak-Szelachowska, M.; Zaczek, M.; Weber-Dabrowska, B.; Miȩdzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóz, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage neutralization by sera of patients receiving phage therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Zaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dabrowska, B.; Miedzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóz, P.; Górski, A. Antibody production in response to staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front. Microbiol. 2016, 7, 1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.Y.; Gill, J.J. Phage therapy redux—What is to be done? Science 2015, 350, 1163–1164. [Google Scholar] [CrossRef] [PubMed]
- Segall, A.M.; Roach, D.R.; Strathdee, S.A. Stronger together? Perspectives on phage-antibiotic synergy in clinical applications of phage therapy. Curr. Opin. Microbiol. 2019, 51, 46–50. [Google Scholar] [CrossRef]
- Williams, H.T.P. Phage-induced diversification improves host evolvability. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Vasse, M.; Torres-Barceló, C.; Hochberg, M.E. Phage selection for bacterial cheats leads to population decline. Proc. R. Soc. B Biol. Sci. 2015, 282. [Google Scholar] [CrossRef] [Green Version]
- Tomich, M.; Herfst, C.A.; Golden, J.W.; Mohr, C.D. Role of flagella in host cell invasion by Burkholderia cepacia. Infect. Immun. 2002, 70, 1799–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage-resistant mutants in yersinia pestis: Identification of phage receptors and attenuation for mice. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.A. Synergistic interaction between phage therapy and antibiotics clears Pseudomonas Aeruginosa infection in endocarditis and reduces virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, A. The mathematics of random mutation and natural selection for multiple simultaneous selection pressures and the evolution of antimicrobial drug resistance. Stat. Med. 2016, 35, 5391–5400. [Google Scholar] [CrossRef]
- Young, B. Mixing new cocktails: Drug interactions in antiretroviral regimens. AIDS Patient Care STDS 2005, 19, 286–297. [Google Scholar] [CrossRef]
- Naghizadeh, M.; Torshizi, M.A.K.; Rahimi, S.; Dalgaard, T.S. Synergistic effect of phage therapy using a cocktail rather than a single phage in the control of severe colibacillosis in quails. Poult. Sci. 2019, 98, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Hesse, S.; Malachowa, N.; Porter, A.R.; Freedman, B.; Kobayashi, S.D.; Gardner, D.J.; Scott, D.P.; Adhya, S.; Deleo, F.R. Bacteriophage treatment rescues mice infected with multidrug-resistant klebsiella pneumoniae st258. mBio 2021, 12. [Google Scholar] [CrossRef]
- Lebeaux, D.; Merabishvili, M.; Caudron, E.; Lannoy, D.; Van Simaey, L.; Duyvejonck, H.; Guillemain, R.; Thumerelle, C.; Podglajen, I.; Compain, F.; et al. A Case of Phage Therapy against Pandrug-Resistant Achromobacter xylosoxidans in a 12-Year-Old Lung-Transplanted Cystic Fibrosis Patient. Viruses 2021, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; De Vos, D.; Friman, V.P.; Pirnay, J.P.; Buckling, A. Effects of sequential and simultaneous applications of bacteriophages on populations of Pseudomonas aeruginosa in vitro and in wax moth larvae. Appl. Environ. Microbiol. 2012, 78, 5646–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, N.; Logan, C.; Yung, G.; Furr, C.L.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef]
- Ryan, E.M.; Alkawareek, M.Y.; Donnelly, R.F.; Gilmore, B.F. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 2012, 65, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, D.; Taha, M.; Yi, Q.L.; Ramirez-Arcos, S.; Diallo, J.S.; Carli, A.; Abdelbary, H. Does treatment order matter? Investigating the ability of bacteriophage to augment antibiotic activity against staphylococcus aureus biofilms. Front. Microbiol. 2018, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akturk, E.; Oliveira, H.; Santos, S.B.; Costa, S.; Kuyumcu, S.; Melo, L.D.R.; Azeredo, J. Synergistic action of phage and antibiotics: Parameters to enhance the killing efficacy against mono and dual-species biofilms. Antibiotics 2019, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.F.; Krisch, H.M. Phage-antibiotic synergy (PAS): β-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Harjai, K.; Chhibber, S. Methicillin-resistant Staphylococcus aureus phage plaque size enhancement using sublethal concentrations of antibiotics. Appl. Environ. Microbiol. 2012, 78, 8227–8233. [Google Scholar] [CrossRef] [Green Version]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex phage-antibiotic synergy (PAS): Antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 2015, 81, 1132–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, J.; Shigehisa, R.; Nasukawa, T.; Mizukami, K.; Takemura-Uchiyama, I.; Ujihara, T.; Murakami, H.; Imanishi, I.; Nishifuji, K.; Sakaguchi, M.; et al. Piperacillin and ceftazidime produce the strongest synergistic phage–antibiotic effect in Pseudomonas aeruginosa. Arch. Virol. 2018, 163, 1941–1948. [Google Scholar] [CrossRef]
- Davis, C.M.; McCutcheon, J.G.; Dennis, J.J. Aztreonam lysine increases the activity of phages e79 and phikz against pseudomonas aeruginosa PA01. Microorganisms 2021, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.; Wahida, A.; Latz, S.; Krüttgen, A.; Häfner, H.; Buhl, E.M.; Ritter, K.; Horz, H.P. Enhanced antibacterial effect of the novel T4-like bacteriophage KARL-1 in combination with antibiotics against multi-drug resistant Acinetobacter baumannii. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef]
- Maiques, E.; Úbeda, C.; Campoy, S.; Salvador, N.; Lasa, Í.; Novick, R.P.; Barbé, J.; Penadés, J.R. β-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 2006, 188, 2726–2729. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jo, Y.; Hwang, Y.J.; Hong, H.W.; Hong, S.S.; Park, K.; Myung, H. Phage-antibiotic synergy via delayed lysis. Appl. Environ. Microbiol. 2018, 84, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Anany, A.M.; Fatima, R.; Hynes, A. Temperate phage-antibiotic synergy eradicates bacteria through depletion of lysogens. Cell Rep. 2021, 35, 109172. [Google Scholar] [CrossRef] [PubMed]
- Nzula, S.; Vandamme, P.; Govan, J.R.W. Sensitivity of the Burkholderia cepacia complex and Pseudomonas aeruginosa to transducing bacteriophages. FEMS Immunol. Med. Microbiol. 2000, 28, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Broxmeyer, L.; Sosnowska, D.; Miltner, E.; Chacoón, O.; Wagner, D.; Mc Garvey, J.; Barletta, R.G.; Bermudez, L.E. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent Mycobacterium: A model for phage therapy of intracellular bacterial pathogens. J. Infect. Dis. 2002, 186, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, K.R.; Clokie, M.R.J. Clostridium difficile phages: Still difficult? Front. Microbiol. 2014, 5, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, R.; Kenna, D.T.; Vandamme, P.; Ure, R.; Govan, J.R.W. Lysogeny and bacteriophage host range within the Burkholderia cepacia complex. J. Med. Microbiol. 2003, 52, 483–490. [Google Scholar] [CrossRef]
- Gill, J.J.; Summer, E.J.; Russell, W.K.; Cologna, S.M.; Carlile, T.M.; Fuller, A.C.; Kitsopoulos, K.; Mebane, L.M.; Parkinson, B.N.; Sullivan, D.; et al. Genomes and characterization of phages Bcep22 and BcepIL02, founders of a novel phage type in Burkholderia cenocepacia. J. Bacteriol. 2011, 193, 5300–5313. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.H.; Stothard, P.; Dennis, J.J. Comparative analysis of two phenotypically-similar but genomically-distinct Burkholderia cenocepacia-specific bacteriophages. BMC Genom. 2012, 13, 223. [Google Scholar] [CrossRef] [Green Version]
- Summer, E.J.; Gonzalez, C.F.; Bomer, M.; Carlile, T.; Embry, A.; Kucherka, A.M.; Lee, J.; Mebane, L.; Morrison, W.C.; Mark, L.; et al. Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J. Bacteriol. 2006, 188, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.H.; Abdu, A.H.; Schobert, M.; Dennis, J.J. Genomic characterization of JG068, a novel virulent podovirus active against Burkholderia cenocepacia. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Goudie, A.D.; Lynch, K.H.; Seed, K.D.; Stothard, P.; Shrivastava, S.; Wishart, D.S.; Dennis, J.J. Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.; Liang, Y.; Eberl, L.; Wishart, D.; Dennis, J. Identification and characterization of ϕH111-1: A novel myovirus with broad activity against clinical isolates of Burkholderia cenocepacia. Bacteriophage 2013, 3, e26649. [Google Scholar] [CrossRef] [Green Version]
- Roszniowski, B.; McClean, S.; Drulis-Kawa, Z. Burkholderia cenocepacia prophages—prevalence, chromosome location and major genes involved. Viruses 2018, 10, 297. [Google Scholar] [CrossRef] [Green Version]
- Summer, E.J.; Gonzalez, C.F.; Carlisle, T.; Mebane, L.M.; Cass, A.M.; Savva, C.G.; LiPuma, J.J.; Young, R. Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 2004, 340, 49–65. [Google Scholar] [CrossRef]
- Golshahi, L.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H. In vitro lung delivery of bacteriophages KS4-M and ΦKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J. Appl. Microbiol. 2011, 110, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Roszniowski, B.; Latka, A.; Maciejewska, B.; Vandenheuvel, D.; Olszak, T.; Briers, Y.; Holt, G.S.; Valvano, M.A.; Lavigne, R.; Smith, D.L.; et al. The temperate Burkholderia phage AP3 of the Peduovirinae shows efficient antimicrobial activity against B. cenocepacia of the IIIA lineage. Appl. Microbiol. Biotechnol. 2017, 101, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Ryley, H.; Jackson, S.K. Highly purified lipopolysaccharides from Burkholderia cepacia complex clinical isolates induce inflammatory cytokine responses via TLR4-mediated MAPK signalling pathways and activation of NFκB. Cell. Microbiol. 2007, 9, 532–543. [Google Scholar] [CrossRef]
- Hutchison, M.L.; Bonell, E.C.; Poxton, I.R.; Govan, J.R.W. Endotoxic activity of lipopolysaccharides isolated from emergent potential cystic fibrosis pathogens. FEMS Immunol. Med. Microbiol. 2000, 27, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.; Poxton, I.R.; Govan, J.R.W. Biological activity of Burkholderia (Pseudomonas) cepacia lipopolysaccharide. FEMS Immunol. Med. Microbiol. 1995, 11, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Golshahi, L.; Seed, K.D.; Dennis, J.J.; Finlay, W.H. Toward modern inhalational bacteriophage therapy: Nebulization of bacteriophages of Burkholderia cepacia complex. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Matinkhoo, S.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H.; Vehring, R. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 2011, 100, 5197–5205. [Google Scholar] [CrossRef]
- Lynch, K.H.; Stothard, P.; Dennis, J.J. Characterization of DC1, a broad-host-range Bcep22-like podovirus. Appl. Environ. Microbiol. 2012, 78, 889–891. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Itoh, Y.; Ohta, S.; Terawaki, Y. A generalized transducing phage of Pseudomonas cepacia. J. Gen. Microbiol. 1986, 132, 2583–2586. [Google Scholar] [CrossRef] [Green Version]
- Hens, D.K.; Ghosh, A.N.; Kumar, R. A new small temperate DNA phage BcP15 isolated from Burkholderia cepacia DR11. Arch. Virol. 2005, 150, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Hens, D.K.; Chatterjee, N.C.; Kumar, R. New temperate DNA phage BcP15 acts as a drug resistance vector. Arch. Virol. 2006, 151, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Weiser, R.; Yap, Z.L.; Otter, A.; Jones, B.V.; Salvage, J.; Parkhill, J.; Mahenthiralingam, E. A novel inducible prophage from Burkholderia vietnamiensis G4 is widely distributed across the species and has lytic activity against pathogenic Burkholderia. Viruses 2020, 12, 601. [Google Scholar] [CrossRef] [PubMed]
- Urban, T.A.; Griffith, A.; Torok, A.M.; Smolkin, M.E.; Burns, J.L.; Goldberg, J.B. Contribution of Burkholderia cenocepacia flagella to infectivity and inflammation. Infect. Immun. 2004, 72, 5126–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjan, U.; Wu, Y.; Kent, G.; Forstner, J. Preferential adherence of cable-piliated Burkholderia cepacia to respiratory epithelia of CF knockout mice and human cystic fibrosis lung explants. J. Med. Microbiol. 2000, 49, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.; Bühler, T.; Weller, P.H.; Brown, M.R.W. Increasing resistance of planktonic and biofilm cultures of Burkholderia cepacia to ciprofloxacin and ceftazidime during exponential growth. J. Antimicrob. Chemother. 1998, 42, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, L.; Cescutti, P.; Drigo, L.; Impallomeni, G.; Herasimenka, Y.; Bevivino, A.; Dalmastri, C.; Tabacchioni, S.; Manno, G.; Zanetti, F.; et al. Exopolysaccharides produced by Burkholderia cenocepacia recA lineages IIIA and IIIB. J. Cyst. Fibros. 2004, 3, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.S.; Silva, I.N.; Oliveira, V.H.; Cunha, R.; Moreira, L.M. Insights into the role of extracellular polysaccharides in Burkholderia adaptation to different environments. Front. Cell. Infect. Microbiol. 2011, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Richau, J.A.; Leitão, J.H.; Correia, M.; Lito, L.; Salgado, M.J.; Barreto, C.; Cescutti, P.; Sá-Correia, I. Molecular typing and exopolysaccharide biosynthesis of Burkholderia cepacia isolates from a Portuguese cystic fibrosis center. J. Clin. Microbiol. 2000, 38, 1651–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.W.; Altman, E.; Beveridge, T.J.; Speert, D.P. Colonial morphology of Burkholderia cepacia complex genomovar III: Implications in exopolysaccharide production, pilus expression, and persistence in the mouse. Infect. Immun. 2003, 71, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, B.A.D.; Chu, K.K.; Bylund, J.; Altman, E.; Speert, D.P. Production of exopolysaccharide by Burkholderia cenocepacia results in altered cell-surface interactions and altered bacterial clearance in mice. J. Infect. Dis. 2004, 190, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seed, K.D.; Dennis, J.J. Development of Galleria mellonella as an alternative infection model for the Burkholderia cepacia complex. Infect. Immun. 2008, 76, 1267–1275. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauman, P.; Dennis, J.J. Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex. Viruses 2021, 13, 1331. https://doi.org/10.3390/v13071331
Lauman P, Dennis JJ. Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex. Viruses. 2021; 13(7):1331. https://doi.org/10.3390/v13071331
Chicago/Turabian StyleLauman, Philip, and Jonathan J. Dennis. 2021. "Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex" Viruses 13, no. 7: 1331. https://doi.org/10.3390/v13071331
APA StyleLauman, P., & Dennis, J. J. (2021). Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex. Viruses, 13(7), 1331. https://doi.org/10.3390/v13071331