
Modular Evolution of Coronavirus Genomes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Nucleotide Sequence Alignments
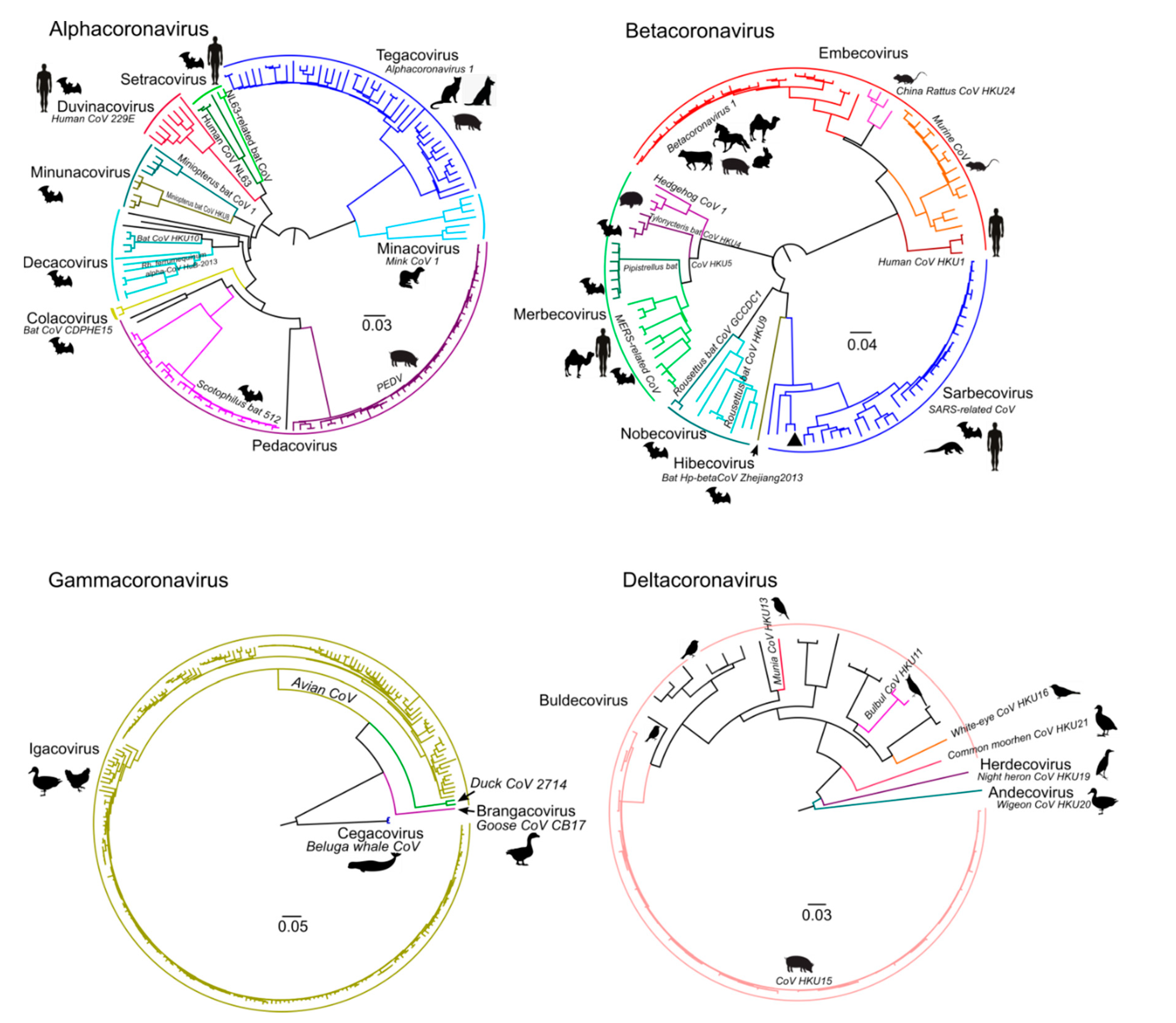
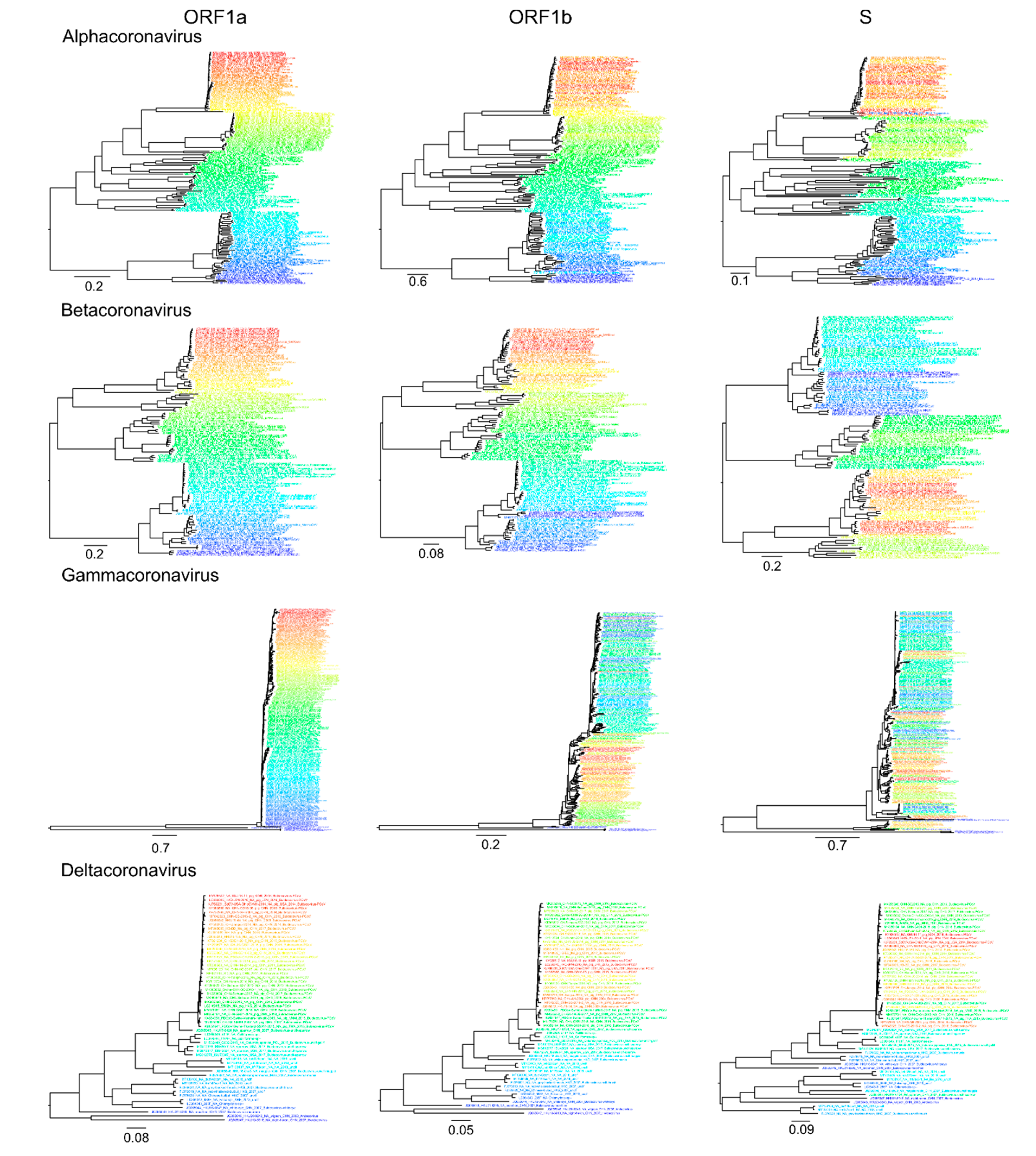
2.2. Phylogenetic Analysis
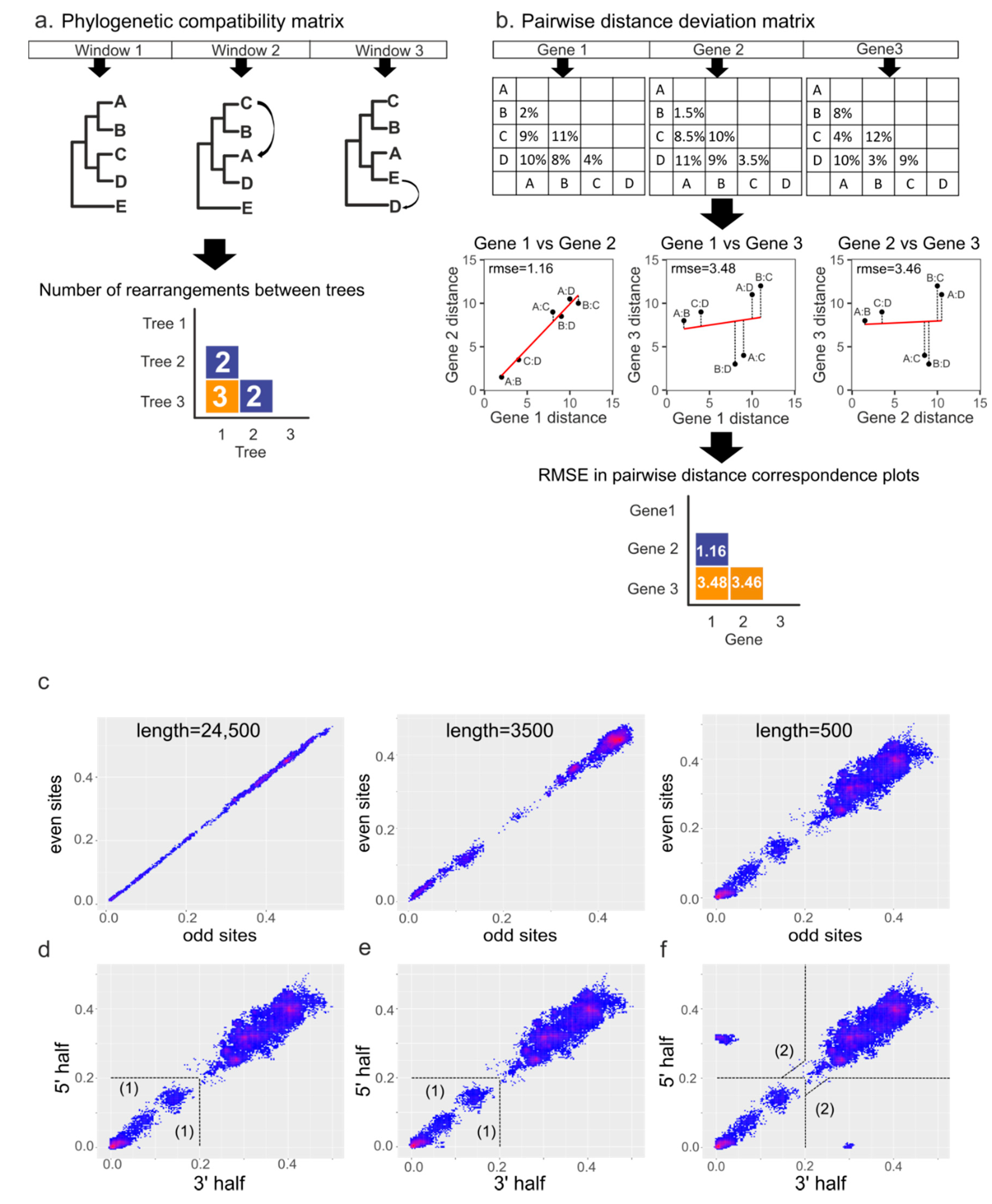
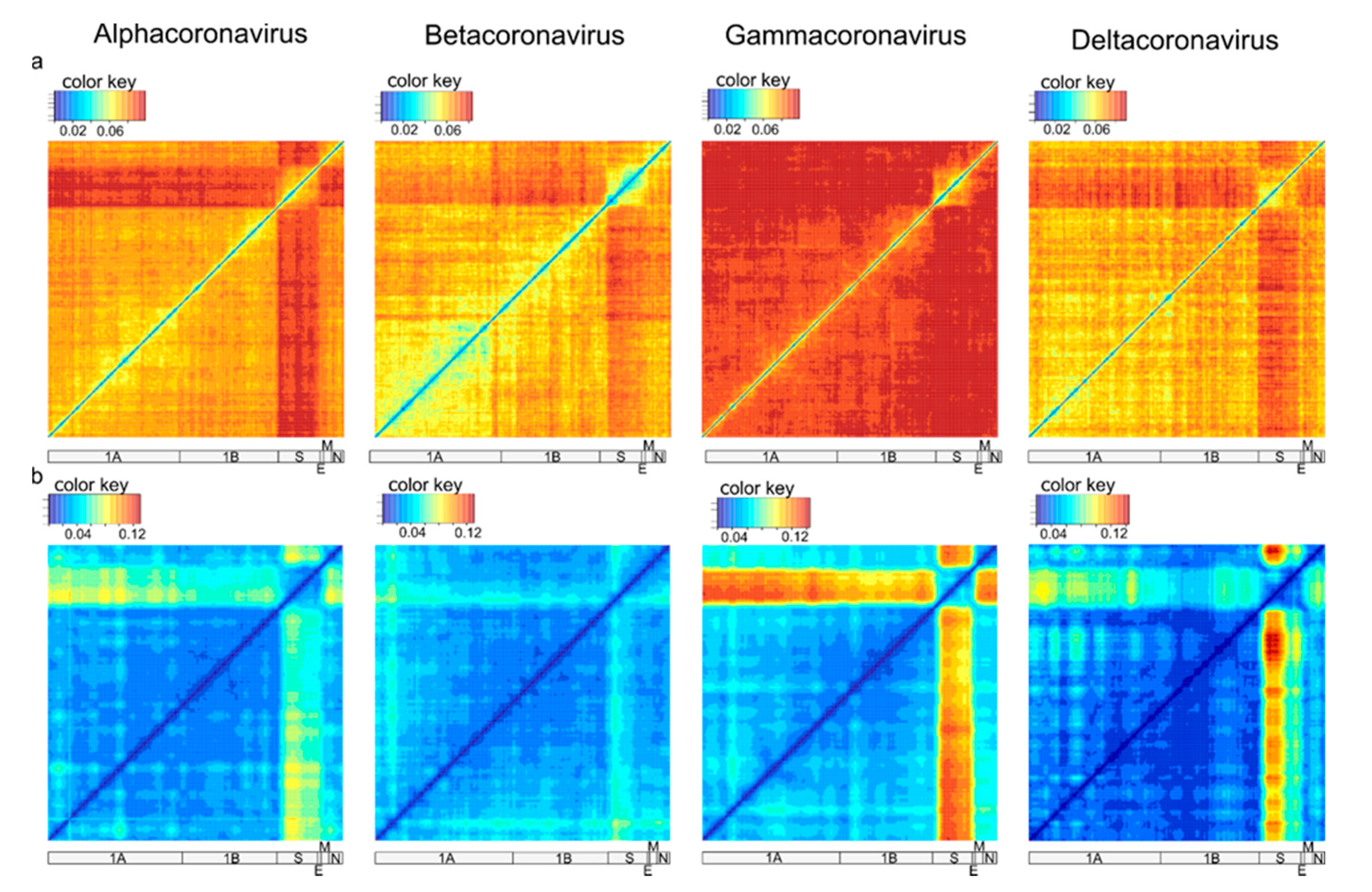
2.3. Recombination Analysis
2.4. Simulation of Recombination Events
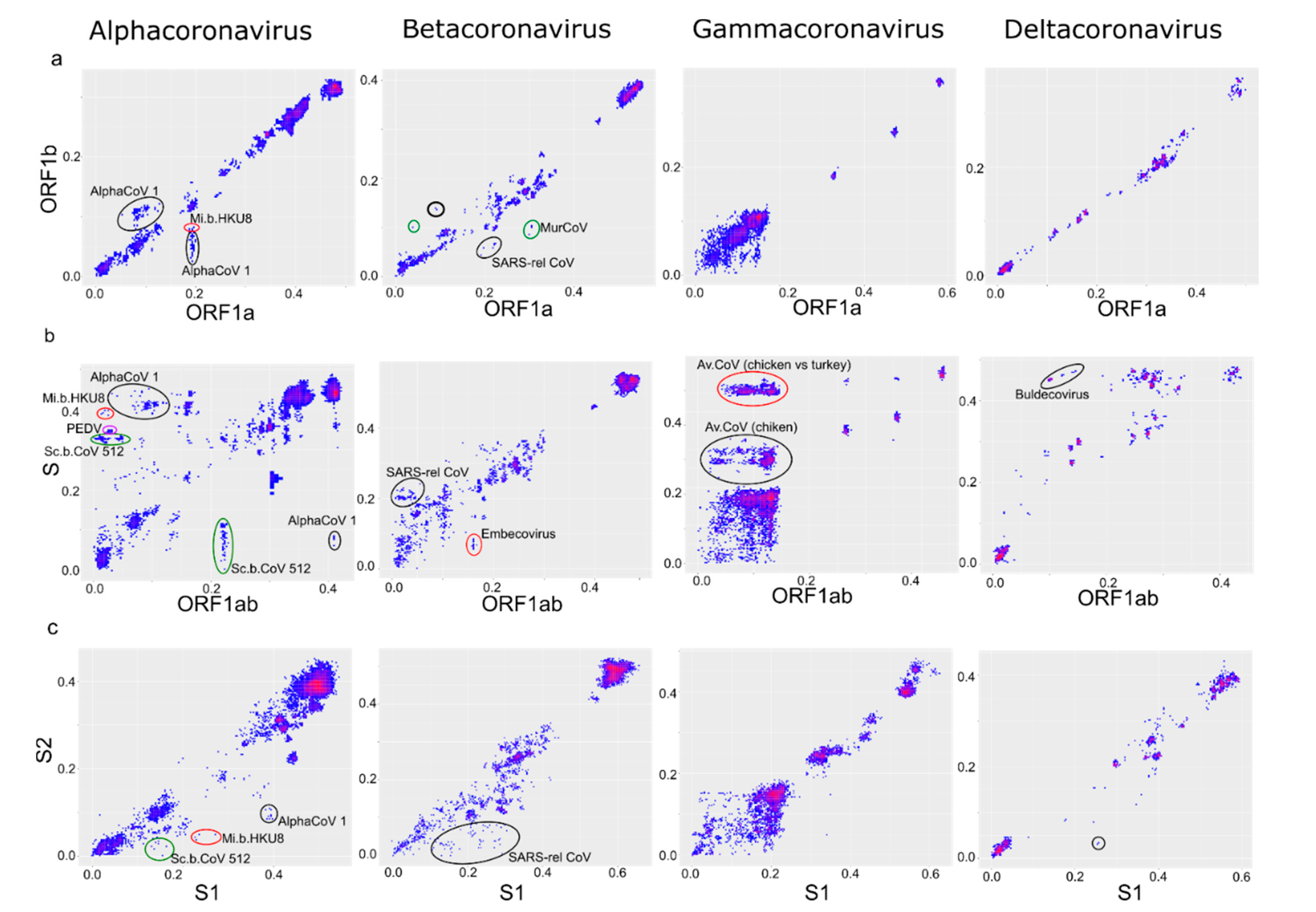
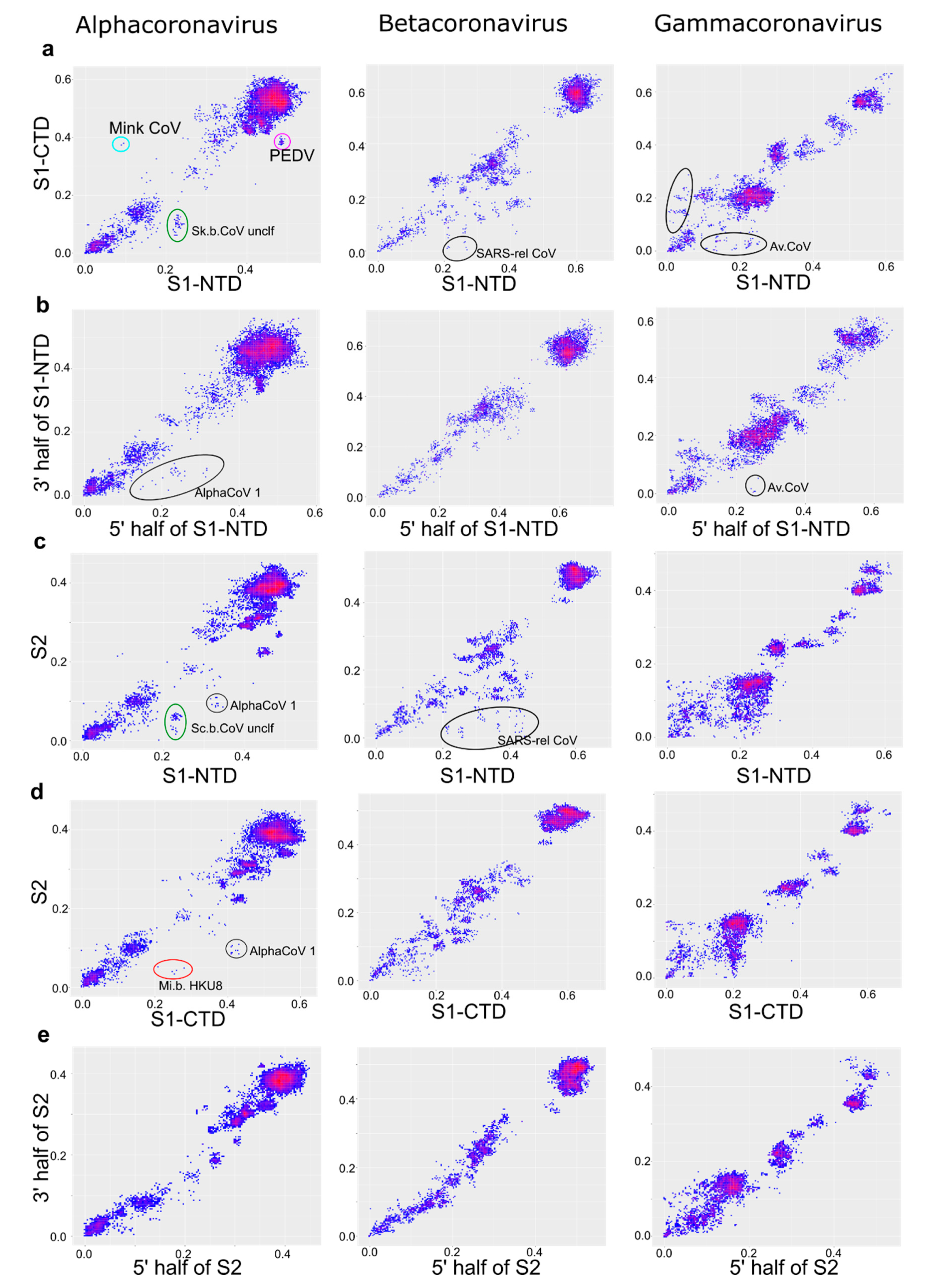
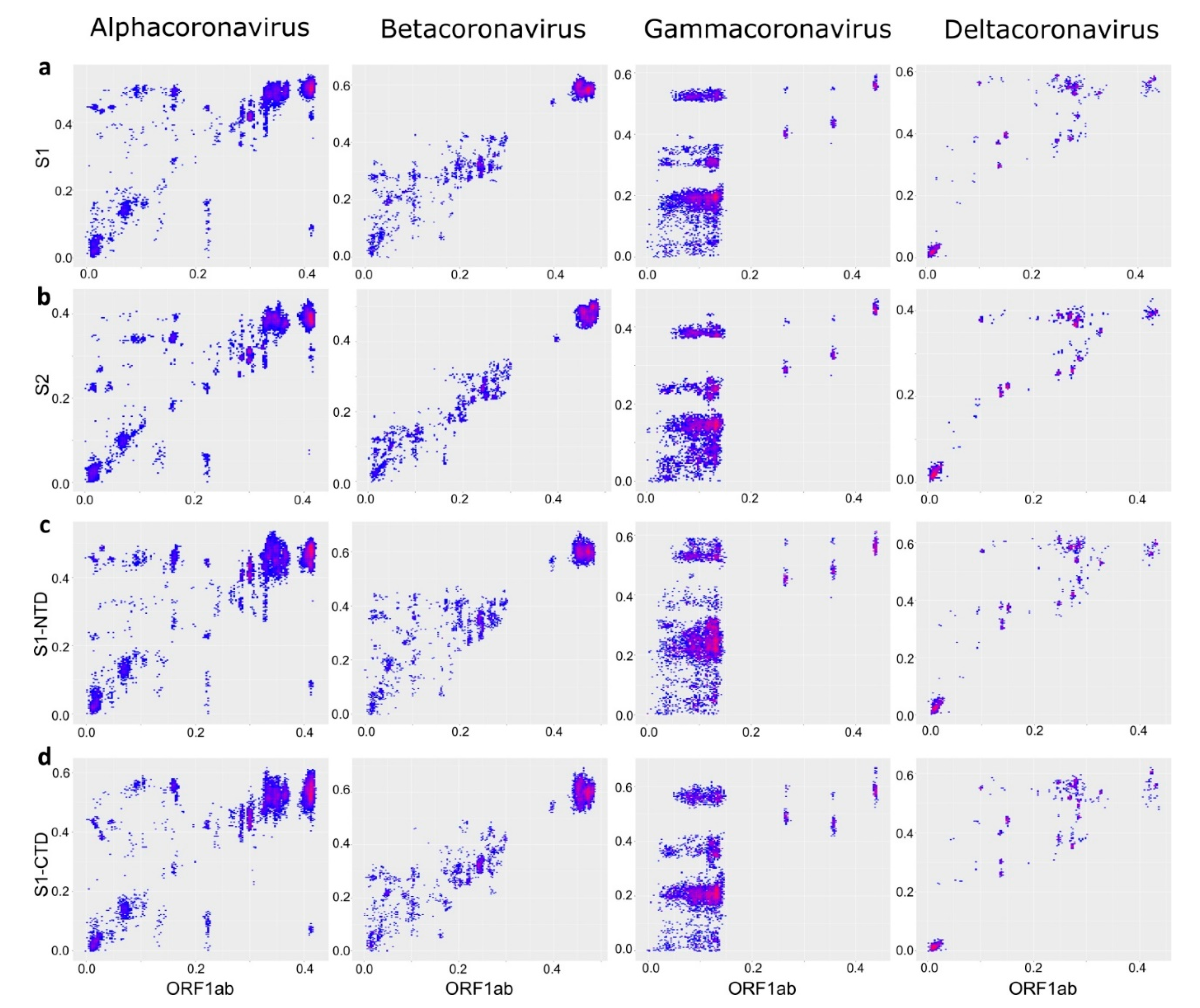
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, W.K.; de Oliveira-Filho, E.F.; Rasche, A.; Greenwood, A.D.; Osterrieder, K.; Drexler, J.F. Potential zoonotic sources of SARS-CoV-2 infections. Transbound. Emerg. Dis. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.-C.; Lam, T.-Y.; Shi, Z.-L.; Drummond, A.J.; Yip, C.-W.; Zeng, F.; Lam, P.-Y.; Leung, F.C.-C. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and Its Implications on the Direct Ancestor of SARS Coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Sabir, J.S.M.; Lam, T.T.-Y.; Ahmed, M.M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.M.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Wille, M.; Holmes, E.C. Wild birds as reservoirs for diverse and abundant gamma- and deltacoronaviruses. FEMS Microbiol. Rev. 2020, 44, 631–644. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; Junglen, S.; et al. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch. Virol. 2019, 164, 2417–2429. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–23. [Google Scholar]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between nonsegmented RNA genomes of murine coronaviruses. J. Virol. 1985, 56, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Corman, V.M.; Schacht, D.; Gloza-Rausch, F.; Seebens-Hoyer, A.; Gmyl, A.P.; Drosten, C.; Drexler, J.F. Close genetic relatedness of picornaviruses from European and Asian bats. J. Gen. Virol. 2017, 98, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Luytjes, W.; Bredenbeek, P.J.; Noten, A.F.H.; Horzinek, M.C.; Spaan, W.J.M. Sequence of mouse hepatitis virus A59 mRNA 2: Indications for RNA recombination between coronaviruses and influenza C virus. Virology 1988, 166, 415–422. [Google Scholar] [CrossRef]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the Phylogenetic Tree of Middle East Respiratory Syndrome Coronavirus by Characterization of a Conspecific Virus from an African Bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, L.; Yang, C.; Zheng, Y.; Gauger, P.C.; Anderson, T.; Harmon, K.M.; Zhang, J.; Yoon, K.-J.; Main, R.G.; et al. The emergence of novel sparrow deltacoronaviruses in the United States more closely related to porcine deltacoronaviruses than sparrow deltacoronavirus HKU17. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Mardani, K.; Noormohammadi, A.H.; Ignjatovic, J.; Browning, G.F. Naturally occurring recombination between distant strains of infectious bronchitis virus. Arch. Virol. 2010, 155, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Kusters, J.G.G.; Jager, E.J.J.; Niesters, H.G.M.G.M.; van der Zeijst, B.A.M. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine 1990, 8, 605–608. [Google Scholar] [CrossRef]
- Herrewegh, A.A.P.M.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.M.; de Groot, R.J. Feline Coronavirus Type II Strains 79-1683 and 79-1146 Originate from a Double Recombination between Feline Coronavirus Type I and Canine Coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef] [Green Version]
- Boniotti, M.B.; Papetti, A.; Lavazza, A.; Alborali, G.; Sozzi, E.; Chiapponi, C.; Faccini, S.; Bonilauri, P.; Cordioli, P.; Marthaler, D. Porcine Epidemic Diarrhea Virus and Discovery of a Recombinant Swine Enteric Coronavirus, Italy. Emerg. Infect. Dis. 2016, 22, 83–87. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Zeng, L.-P.; Yang, X.-L.; Ge, X.-Y.; Zhang, W.; Li, B.; Xie, J.-Z.; Shen, X.-R.; Zhang, Y.-Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree 1.4.4. Available online: https://github.com/rambaut/figtree/releases (accessed on 1 June 2019).
- Simmonds, P.; Welch, J. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, I.B.; Easteal, S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Bioinformatics 1996, 12, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A.N.; Shumilina, E.Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Deviatkin, A.A.; Lukashev, A.N. Recombination in the rabies virus and other lyssaviruses. Infect. Genet. Evol. 2018, 60, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Midgley, S. Recombination in the Genesis and Evolution of Hepatitis B Virus Genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Bi, Y.; Liu, J.; Zhou, J.; Liu, W.; Lai, A.C.K.; Wong, G.; Shi, W.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Chu, D.K.W.; Peiris, J.S.M.; Kosakovsky Pond, S.L.; Poon, L.L.M. A Case for the Ancient Origin of Coronaviruses. J. Virol. 2013, 87, 7039–7045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baric, R.S.; Fu, K.; Schaad, M.C.; Stohlman, S.A. Establishing a genetic recombination map for murine coronavirus strain A59 complementation groups. Virology 1990, 177, 646–656. [Google Scholar] [CrossRef]
- Banner, L.R.; Mc Lai, M. Random nature of coronavirus RNA recombination in the absence of selection pressure. Virology 1991, 185, 441–445. [Google Scholar] [CrossRef]
- Fu, K.; Baric, R.S. Map locations of mouse hepatitis virus temperature-sensitive mutants: Confirmation of variable rates of recombination. J. Virol. 1994, 68, 7458–7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesley, R.D. The S gene of canine coronavirus, strain UCD-1, is more closely related to the S gene of transmissible gastroenteritis virus than to that of feline infectious peritonitis virus. Virus Res. 1999, 61, 145–152. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, R.T.Y.; Chu, D.K.W.; Miguel, E.; Perera, R.A.P.M.; Oladipo, J.O.; Fassi-Fihri, O.; Aylet, G.; Ko, R.L.W.; Zhou, Z.; Cheng, M.-S.; et al. Diversity of Dromedary Camel Coronavirus HKU23 in African Camels Revealed Multiple Recombination Events among Closely Related Betacoronaviruses of the Subgenus Embecovirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of 22 Coronavirus HKU1 Genomes Reveals a Novel Genotype and Evidence of Natural Recombination in Coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Yang, J.; Lu, S.; Lan, R.; Jin, D.; Luo, X.; Pu, J.; Wu, S.; Xu, J. Beta- and Novel Delta-Coronaviruses Are Identified from Wild Animals in the Qinghai-Tibetan Plateau, China. Virol. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Fan, R.Y.Y.; Lam, C.S.F.; Li, K.S.M.; Ahmed, S.S.; Chow, F.W.N.; Cai, J.-P.; Zhu, X.; et al. Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China. Viruses 2019, 11, 980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinteros, J.A.; Lee, S.-W.; Markham, P.F.; Noormohammadi, A.H.; Hartley, C.A.; Legione, A.R.; Coppo, M.J.C.; Vaz, P.K.; Browning, G.F. Full genome analysis of Australian infectious bronchitis viruses suggests frequent recombination events between vaccine strains and multiple phylogenetically distant avian coronaviruses of unknown origin. Vet. Microbiol. 2016, 197, 27–38. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.-J. Origin, Evolution, and Genotyping of Emergent Porcine Epidemic Diarrhea Virus Strains in the United States. MBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, Y.; Zhu, X.; Shi, H.; Chen, J.; Shi, D.; Yuan, J.; Cao, L.; Liu, J.; Dong, H.; et al. Identification of a natural recombinant transmissible gastroenteritis virus between Purdue and Miller clusters in China. Emerg. Microbes Infect. 2017, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular Epidemiology of Human Coronavirus OC43 Reveals Evolution of Different Genotypes over Time and Recent Emergence of a Novel Genotype due to Natural Recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escutenaire, S.; Isaksson, M.; Renström, L.H.M.; Klingeborn, B.; Buonavoglia, C.; Berg, M.; Belák, S.; Thorén, P. Characterization of divergent and atypical canine coronaviruses from Sweden. Arch. Virol. 2007, 152, 1507–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant Canine Coronaviruses Related to Transmissible Gastroenteritis Virus of Swine Are Circulating in Dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Alanis, A.; Sandner-Miranda, L.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. The receptor binding domain of SARS-CoV-2 spike protein is the result of an ancestral recombination between the bat-CoV RaTG13 and the pangolin-CoV MP789. BMC Res. Notes 2020, 13, 398. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yap, Y.L.; Danchin, A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch. Virol. 2005, 150, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Li, F. Receptor Recognition Mechanisms of Coronaviruses: A Decade of Structural Studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef] [Green Version]
- McWilliam Leitch, E.C.; Bendig, J.; Cabrerizo, M.; Cardosa, J.; Hyypia, T.; Ivanova, O.E.; Kelly, A.; Kroes, A.C.M.; Lukashev, A.; MacAdam, A.; et al. Transmission Networks and Population Turnover of Echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef] [Green Version]
- Dudas, G.; Rambaut, A. MERS-CoV recombination: Implications about the reservoir and potential for adaptation. Virus Evol. 2016, 2, vev023. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Number of Sequences in the Dataset | Length of the Alignment, nt |
|---|---|---|
| Alphacoronavirus | 164 | 24,585 |
| Betacoronavirus | 122 | 24,690 |
| Gammacoronavirus | 260 | 24,870 |
| Deltacoronavirus | 56 | 23,840 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. https://doi.org/10.3390/v13071270
Vakulenko Y, Deviatkin A, Drexler JF, Lukashev A. Modular Evolution of Coronavirus Genomes. Viruses. 2021; 13(7):1270. https://doi.org/10.3390/v13071270
Chicago/Turabian StyleVakulenko, Yulia, Andrei Deviatkin, Jan Felix Drexler, and Alexander Lukashev. 2021. "Modular Evolution of Coronavirus Genomes" Viruses 13, no. 7: 1270. https://doi.org/10.3390/v13071270
APA StyleVakulenko, Y., Deviatkin, A., Drexler, J. F., & Lukashev, A. (2021). Modular Evolution of Coronavirus Genomes. Viruses, 13(7), 1270. https://doi.org/10.3390/v13071270