
Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
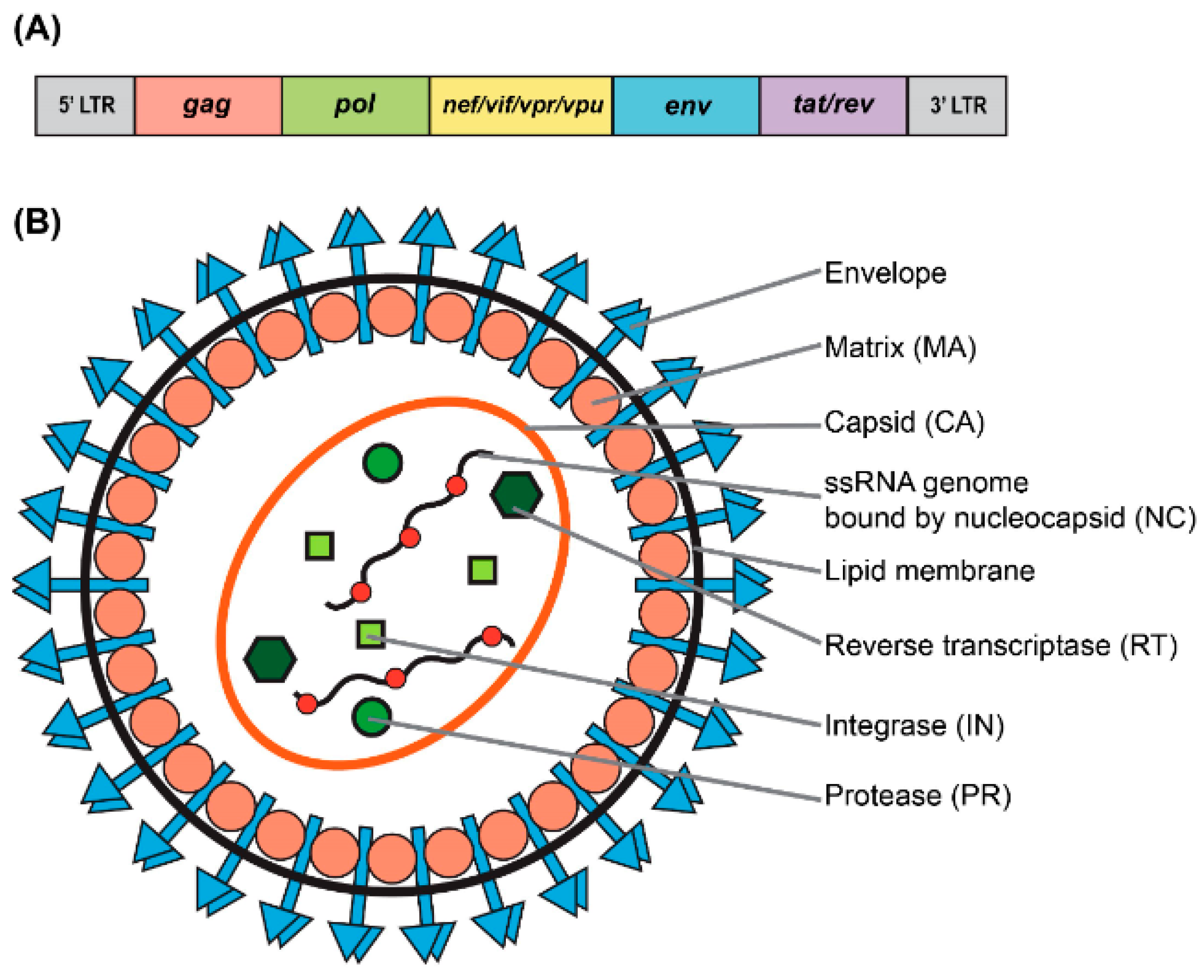
:1. HIV-1 Based Vectors: Basic Biology
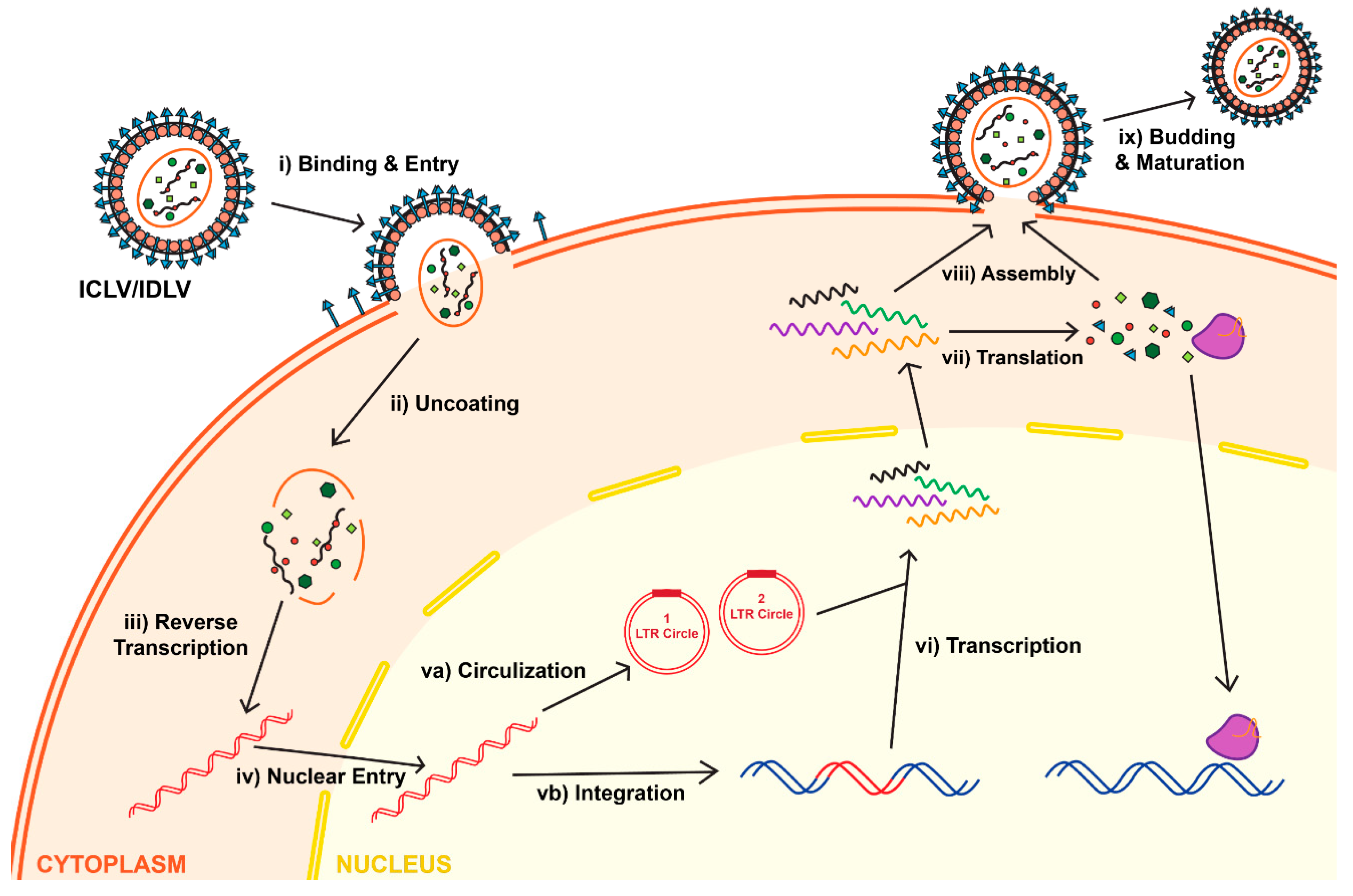
2. The Life Cycle of Lentiviral Vectors
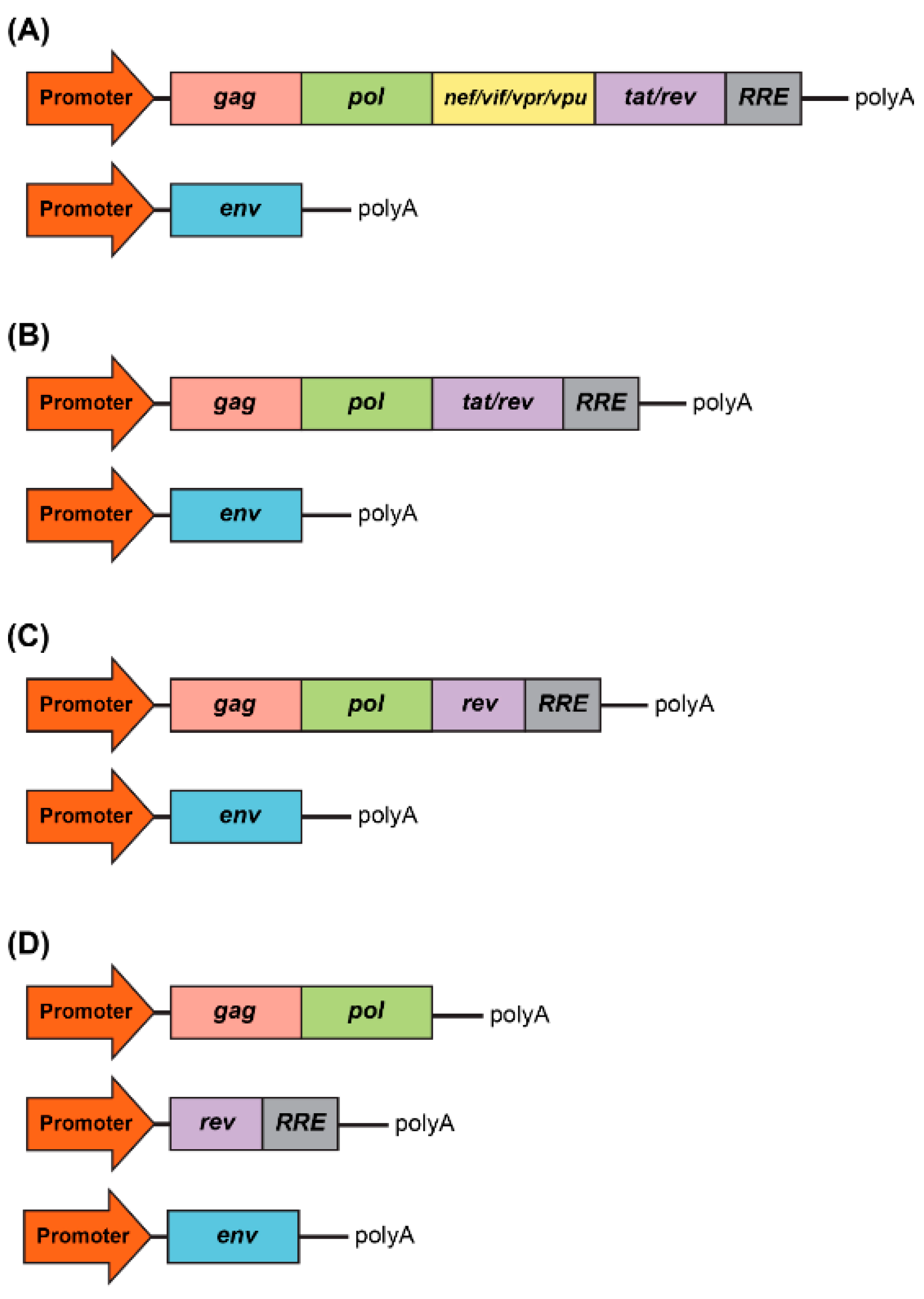
3. Safety of Lentiviral Vectors
4. Non-Integrating Lentiviral Vectors
5. Adeno-Associate Vectors (AAVs)
6. Overview of CRISPR/Cas9-Based Gene-Editing Systems
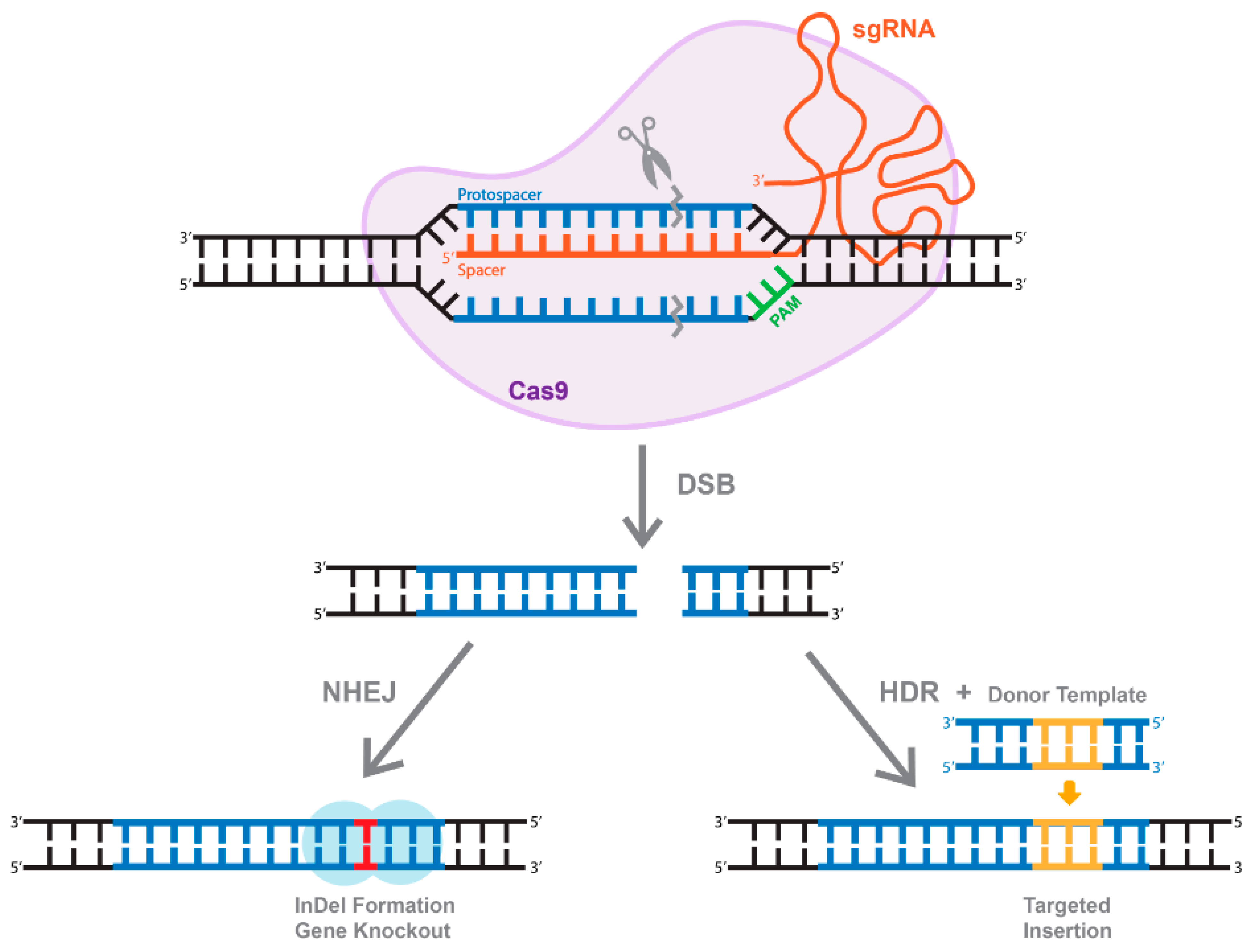
7. The Use of Active Cas9 for Genome-Editing Applications
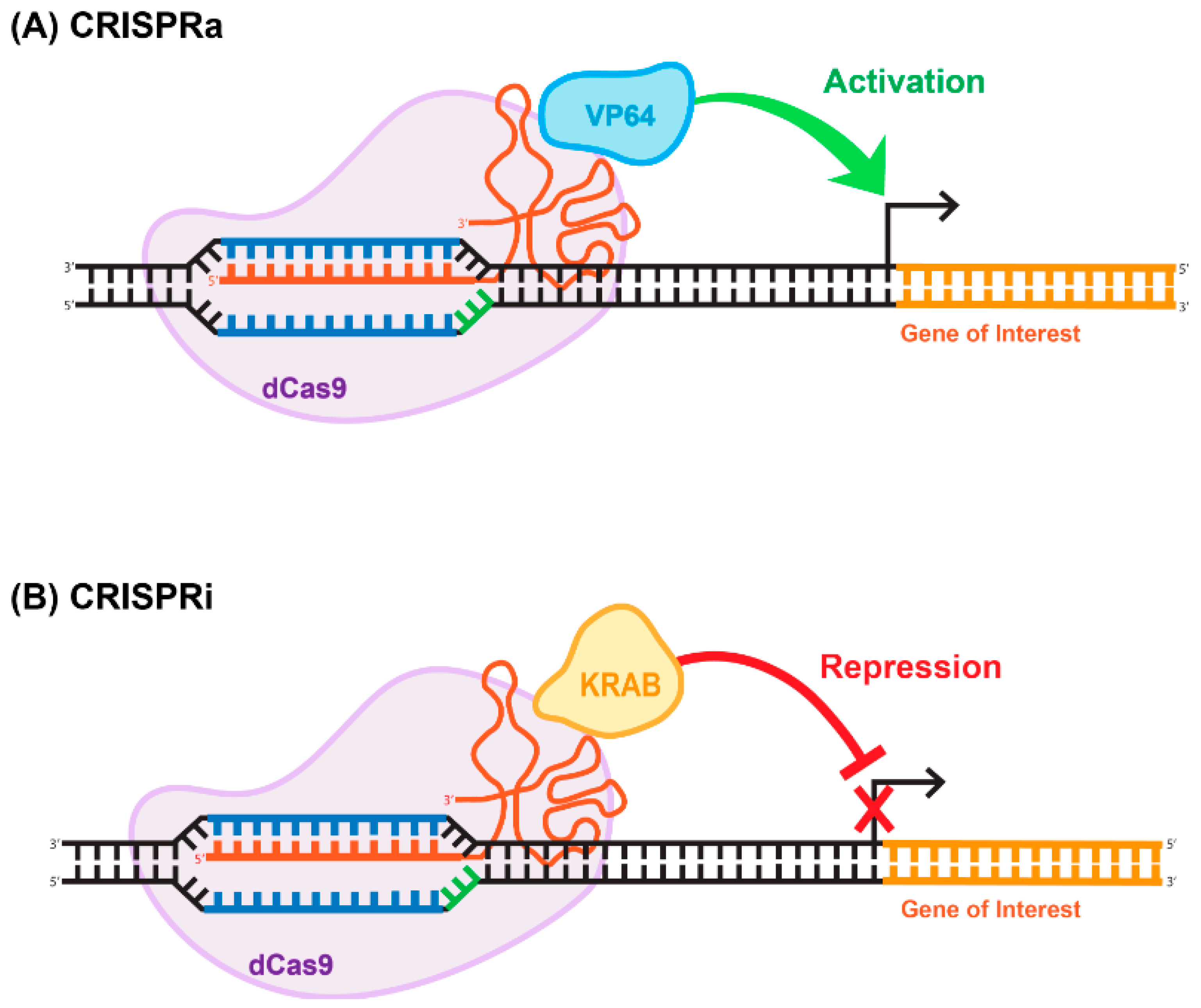
8. Deactivated Cas9 (dCas9) for CRISPRi and CRISPRa Approaches
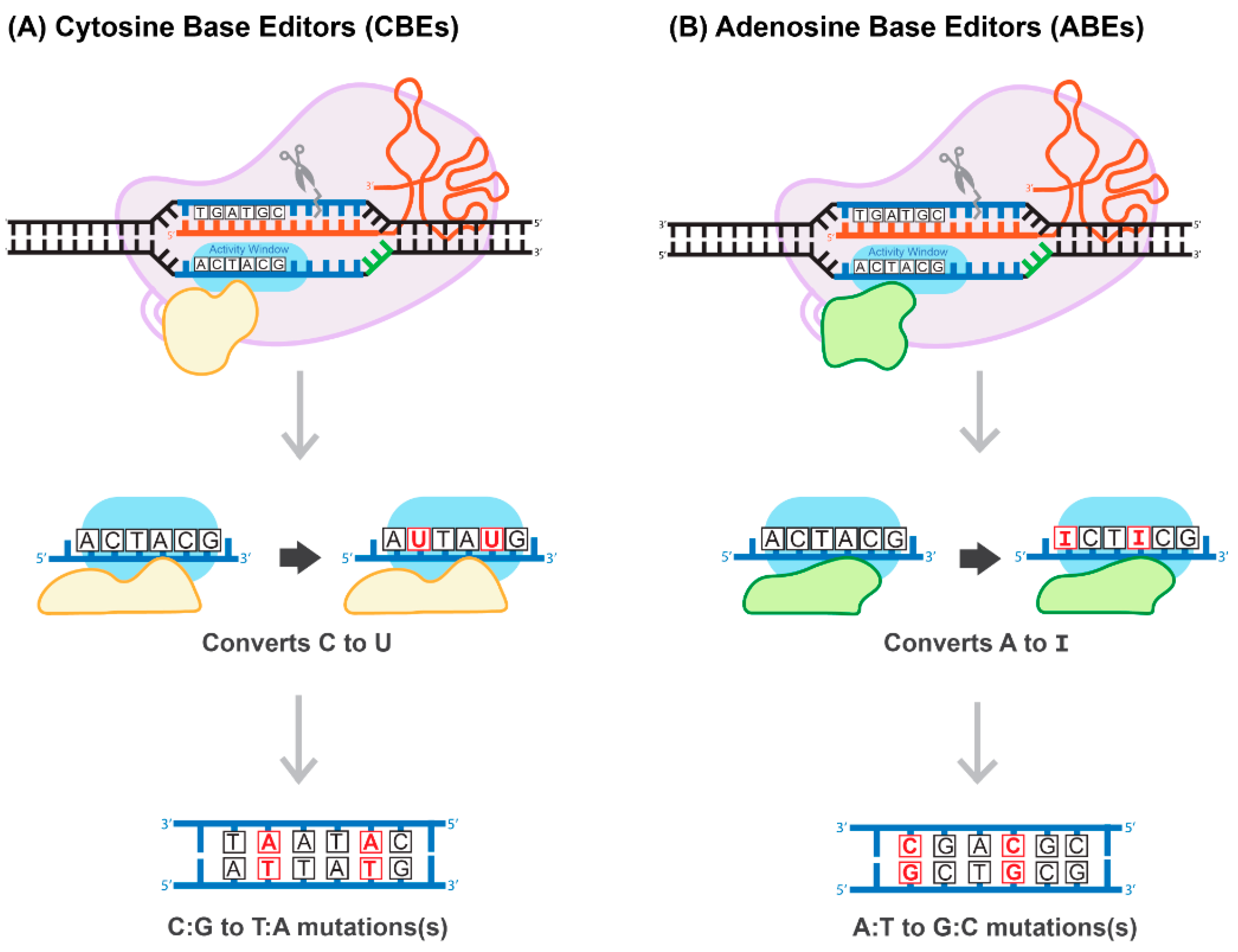
9. Base-Pair Editing Technology
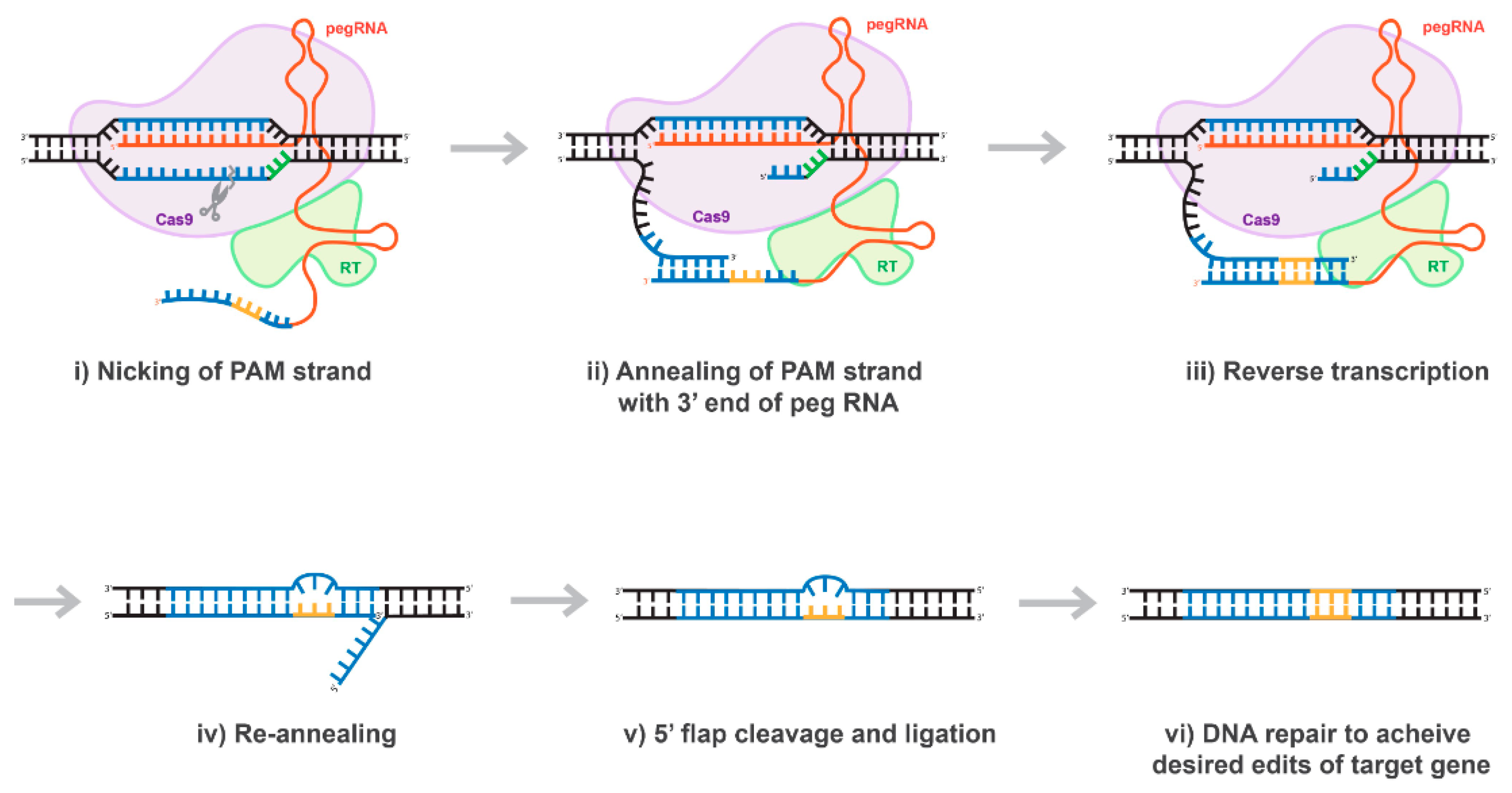
10. Prime-Editing Technology
11. Lentiviral Vectors Paired with Genome-Editing Tools
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Bailey, R.M.; Wimberly, K.; Kalburgi, S.N.; Gray, S.J. Methods for Gene Transfer to the Central Nervous System. Agric. Food Prod. 2014, 87, 125–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Reiser, J. Altering the Tropism of Lentiviral Vectors through Pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. The Interactions of Retroviruses and their Hosts. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomer, U.; Naldini, L.; Kafri, T.; Trono, D.; Verma, I.M.; Gage, F.H. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J. Virol. 1997, 71, 6641–6649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafri, T.; Blömer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, A.S.; Ma, H.; Fu, K.; McCown, T.J.; Kafri, T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Mol. Ther. 2006, 14, 276–284. [Google Scholar] [CrossRef]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [CrossRef] [Green Version]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 Genome Nuclear Import Is Mediated by a Central DNA Flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J.; Goff, S.P. Mutants and pseudorevertants of moloney murine leukemia virus with alterations at the integration site. Cell 1985, 42, 573–580. [Google Scholar] [CrossRef]
- Craigie, R.; Fujiwara, T.; Bushman, F. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 1990, 62, 829–837. [Google Scholar] [CrossRef]
- Leavitt, A.D.; Rose, R.B.; Varmus, H.E. Both substrate and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced in Saccharomyces cerevisiae. J. Virol. 1992, 66, 2359–2368. [Google Scholar] [CrossRef] [Green Version]
- Buchow, H.D.; Tschachler, E.; Gallo, R.C.; Reitz, M. HIV-I Replication Requires an Intact Integrase Reading Frame. Acute Leuk. 1989, 32, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.N.; Mitrophanous, K.; Kingsman, S.M.; Kingsman, A.J. Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J. Virol. 1998, 72, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Blomer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, T.; Foley, P.; Wirth, M.; Munch, J.; Uberla, K. Development of a Self-Inactivating, Minimal Lentivirus Vector Based on Simian Immunodeficiency Virus. Hum. Gene Ther. 2000, 11, 439–447. [Google Scholar] [CrossRef]
- Iwakuma, T.; Cui, Y.; Chang, L.-J. Self-Inactivating Lentiviral Vectors with U3 and U5 Modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Ma, H.; Webster-Cyriaque, J.; Monahan, P.E.; Kafri, T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc. Natl. Acad. Sci. USA 2009, 106, 18786–18791. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; Basile, G.D.S.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; DI Serio, M.S.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 2021, 29, 489–504. [Google Scholar] [CrossRef]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Schuessler-Lenz, M.; Enzmann, H.; Vamvakas, S. Regulators’ Advice Can Make a Difference: European Medicines Agency Approval of Zynteglo for Beta Thalassemia. Clin. Pharmacol. Ther. 2020, 107, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, M.; Kantor, B.; Cockrell, A.; Ma, H.; Zeithaml, B.; Li, X.; McCown, T.; Kafri, T. A Large U3 Deletion Causes Increased In Vivo Expression From a Nonintegrating Lentiviral Vector. Mol. Ther. 2008, 16, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Muñoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Philippe, S.; Sarkis, C.; Barkats, M.; Mammeri, H.; Ladroue, C.; Petit, C.; Mallet, J.; Serguera, C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 17684–17689. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Bayer, M.; Ma, H.; Samulski, J.; Li, C.; McCown, T.J.; Kafri, T. Notable Reduction in Illegitimate Integration Mediated by a PPT-deleted, Nonintegrating Lentiviral Vector. Mol. Ther. 2011, 19, 547–556. [Google Scholar] [CrossRef]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. - Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Saida, H.; Matsuzaki, Y.; Takayama, K.; Iizuka, A.; Konno, A.; Yanagi, S.; Hirai, H. One-year follow-up of transgene expression by integrase-defective lentiviral vectors and their therapeutic potential in spinocerebellar ataxia model mice. Gene Ther. 2014, 21, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VijayRaghavan, S.; Kantor, B. A Protocol for the Production of Integrase-deficient Lentiviral Vectors for CRISPR/Cas9-mediated Gene Knockout in Dividing Cells. J. Vis. Exp. 2017, e56915. [Google Scholar] [CrossRef]
- Kantor, B.; McCown, T.; Leone, P.; Gray, S.J. Clinical Applications Involving CNS Gene Transfer. Adv. Genet. 2014, 87, 71–124. [Google Scholar] [CrossRef] [Green Version]
- Rittiner, J.E.; Moncalvo, M.; Chiba-Falek, O.; Kantor, B. Gene-Editing Technologies Paired With Viral Vectors for Translational Research Into Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 148. [Google Scholar] [CrossRef]
- Lim, C.K.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; C., M.A.Z.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR — A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Genet. 2008, 6, 181–186. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Annotation and Classification of CRISPR-Cas Systems. Methods Mol. Biol. 2015, 1311, 47–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.; Barrangou, R.; Brouns, S.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Genet. 2015, 13, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nat. Cell Biol. 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Hsu, P.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Dammes, N.; Peer, D. Progress and challenges towards CRISPR/Cas clinical translation. Adv. Drug Deliv. Rev. 2020, 154-155, 176–186. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. Editing the Genome Without Double-Stranded DNA Breaks. ACS Chem. Biol. 2018, 13, 383–388. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.; Levy, J.M.; Chen, P.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nat. Cell Biol. 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.; Habib, N.; Gootenberg, J.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nat. Cell Biol. 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.D.; Pruitt, B.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nat. Cell Biol. 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, L.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nat. Cell Biol. 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjana, N.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.; Garcia, S.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nat. Cell Biol. 2015, 527, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene. Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.V.; Hollis, R.P.; Kuftinec, G.; Senadheera, S.; Chan, R.; Kohn, D.B. Integrase-defective Lentiviral Vectors as a Delivery Platform for Targeted Modification of Adenosine Deaminase Locus. Mol. Ther. 2013, 21, 1705–1717. [Google Scholar] [CrossRef] [Green Version]
- Tagliafierro, L.; Ilich, E.; Moncalvo, M.; Gu, J.; Sriskanda, A.; Grenier, C.; Murphy, S.K.; Chiba-Falek, O.; Kantor, B. Lentiviral Vector Platform for the Efficient Delivery of Epigenome-editing Tools into Human Induced Pluripotent Stem Cell-derived Disease Models. J. Vis. Exp. 2019, e59241. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. https://doi.org/10.3390/v13071288
Dong W, Kantor B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses. 2021; 13(7):1288. https://doi.org/10.3390/v13071288
Chicago/Turabian StyleDong, Wendy, and Boris Kantor. 2021. "Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives" Viruses 13, no. 7: 1288. https://doi.org/10.3390/v13071288
APA StyleDong, W., & Kantor, B. (2021). Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses, 13(7), 1288. https://doi.org/10.3390/v13071288