
Intracellular Sequestration of the NKG2D Ligand MIC B by Species F Adenovirus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Growth and Cells
2.2. Immunofluorescence Staining
2.3. Flow Cytometry
2.4. Protein Sequence Analysis
3. Results
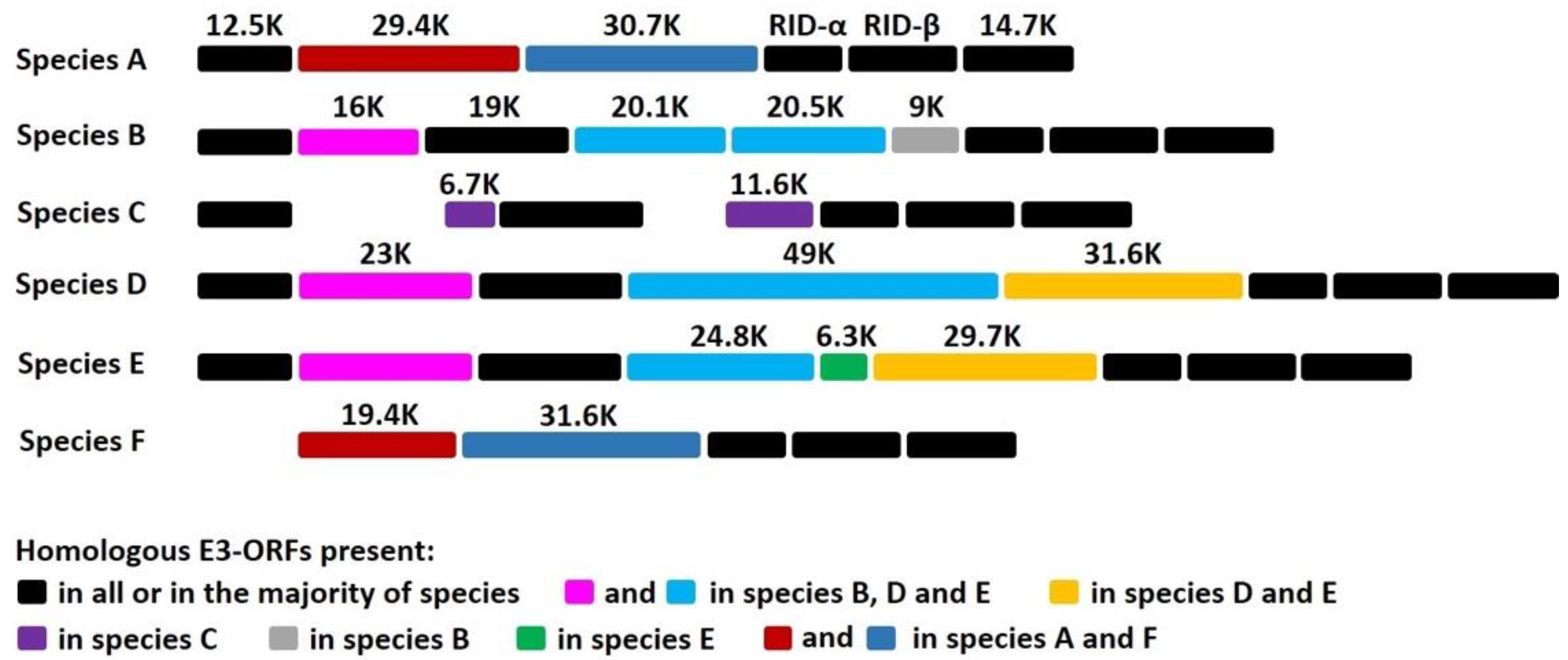
3.1. E3 Region
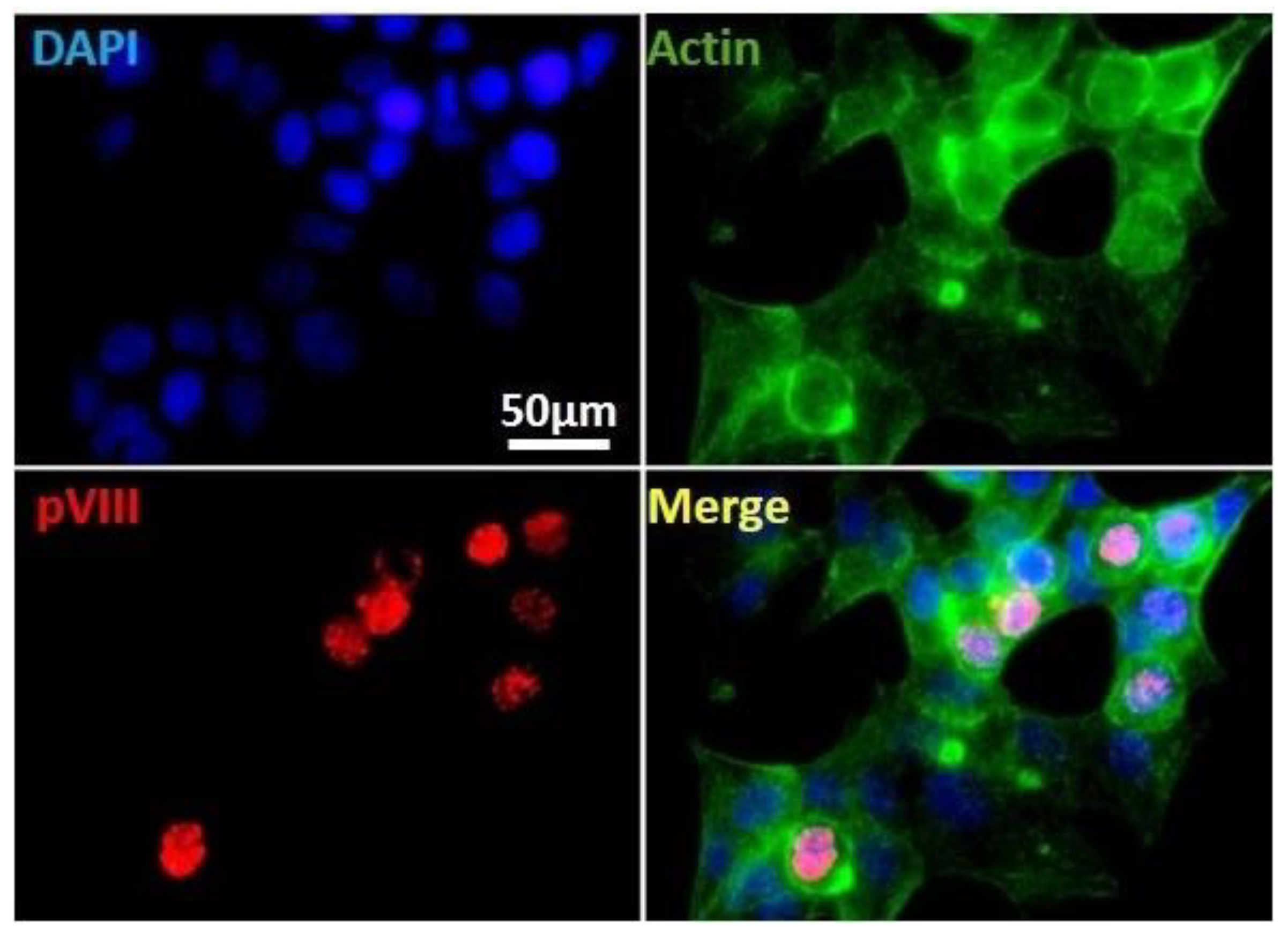
3.2. In Vitro Models of HAdV-F Infection
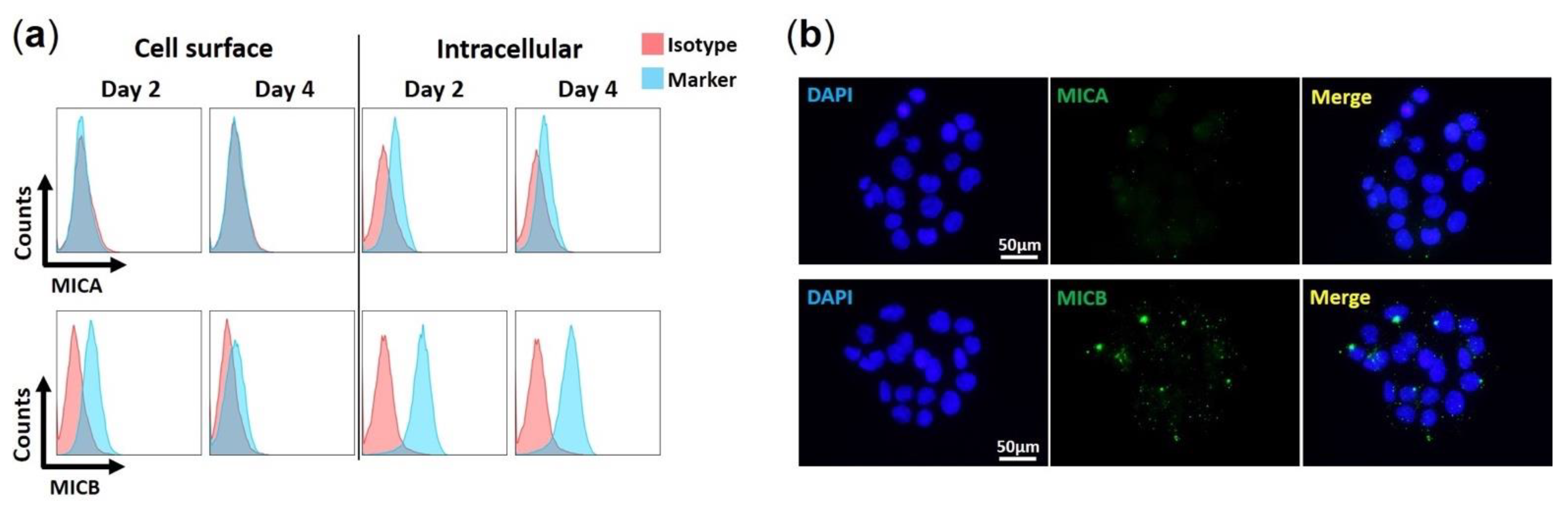
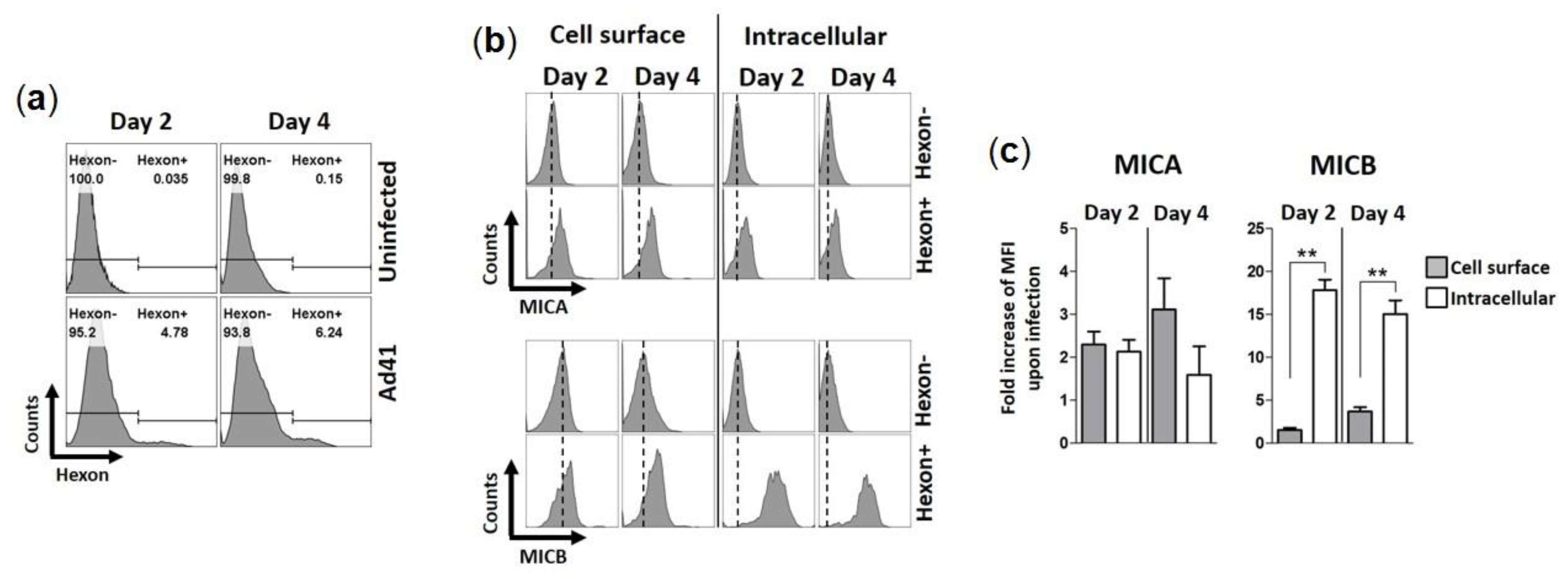
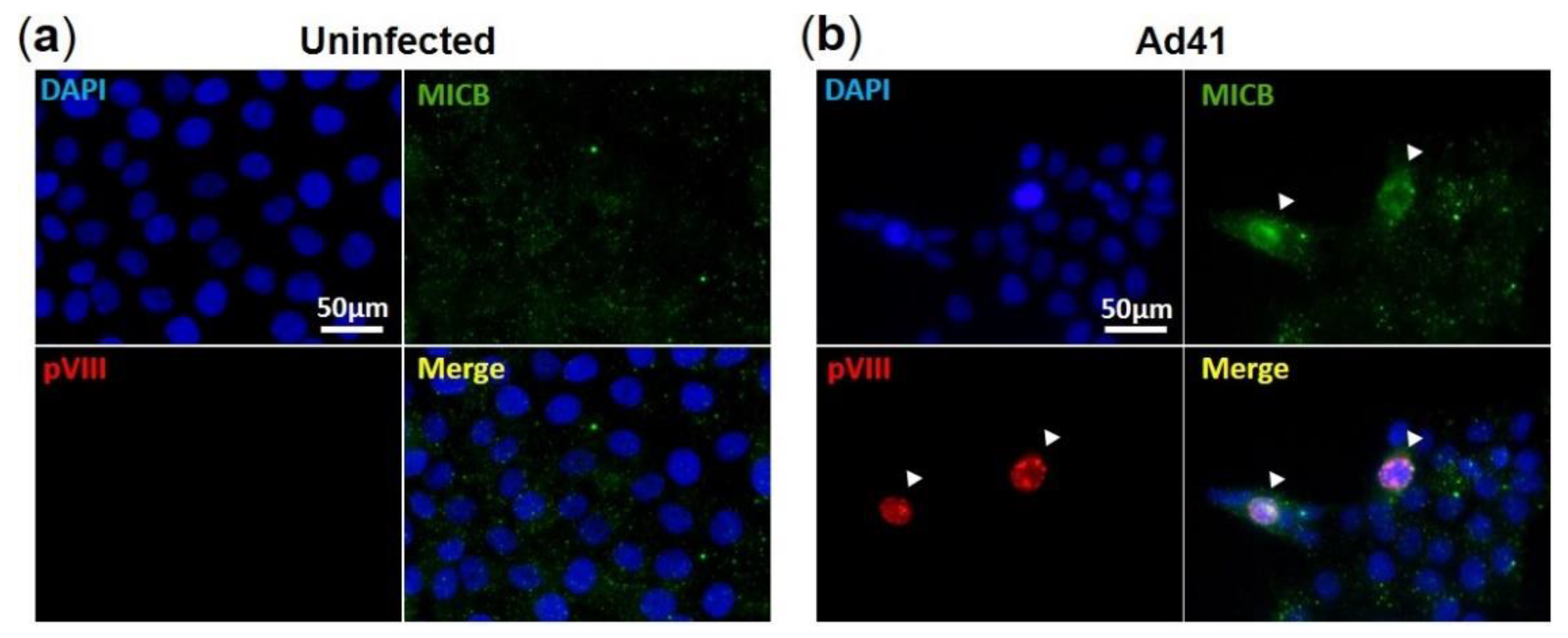
3.3. HAdV-F41 Interferes with Cell Surface Expression of MIC B
3.4. E3-19.4K and E3-31.6K Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berk, A. Adenoviridae. In Fields Virology, 6th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1704–1731. [Google Scholar]
- Lion, T. Adenovirus infections in immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.A. Adenovirus infections in solid organ transplant recipients. Curr. Opin. Organ. Transplant. 2009, 14, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Feghoul, L.; Chevret, S.; Cuinet, A.; Dalle, J.H.; Ouachee, M.; Yacouben, K.; Fahd, M.; Guerin-El Khourouj, V.; Roupret-Serzec, J.; Sterkers, G.; et al. Adenovirus infection and disease in paediatric haematopoietic stem cell transplant patients: Clues for antiviral preemptive treatment. Clin. Microbiol. Infect. 2015, 21, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.T.; Boge, C.L.K.; Petersen, H.; Seif, A.E.; Bryan, M.; Hodinka, R.L.; Cardenas, A.M.; Purdy, D.R.; Loudon, B.; Kajon, A.E. Outcomes of human adenovirus infection and disease in a retrospective cohort of pediatric hematopoietic cell transplant patients. J. Pediatr. Infect. Dis. Soc. 2018, 8, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Burgert, H.; Blusch, J.H. Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 2000, 21, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.M.; Gooding, L.R. Region E3 of adenovirus: A cassette of genes involved in host immunosurveillance and virus-cell interactions. Virology 1991, 184, 1–8. [Google Scholar] [CrossRef]
- Burgert, H.-G.; Kvist, S. The E3/19K protein of adenovirus type 2 binds to the domains of histocompatibility antigens required for CTL recognition. EMBO J. 1987, 6, 2019–2026. [Google Scholar] [CrossRef]
- Burgert, H.-G.; Kvist, S. An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 1985, 41, 987–997. [Google Scholar] [CrossRef]
- Kvist, S.; Ostberg, L.; Persson, H.; Philipson, L.; Peterson, P.A. Molecular association between transplantation antigens and cell surface antigen in adenovirus-transformed cell line. Proc. Natl. Acad. Sci. USA 1978, 75, 5674–5678. [Google Scholar] [CrossRef] [Green Version]
- Paabo, S.; Bhat, B.M.; Wold, W.S.M.; Peterson, P.A. A short sequence in the COOH terminus makes an adenovirus membrane glycoprotein a resident of the endoplasmic reticulum. Cell 1987, 50, 311–317. [Google Scholar] [CrossRef]
- Sester, M.; Ruszics, Z.; Mackley, E.; Burgert, H.-G. The transmembrane domain of the adenovirus E3/19K protein acts as an endoplasmic reticulum retention signal and contribute to intracellular sequestration of major histocompatibility complex class I molecules. J. Virol. 2013, 87, 6104–6117. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.H.; Bennink, J.R.; Yewdell, J.W. Retention of adenovirus E19 glycoprotein in the endoplasmic reticulum is essential to its ability to block antigen presentation. J. Exp. Med. 1991, 174, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Stafford, W.F.; Bouvier, M. The endoplasmic reticulum lumenal domain of the adenovirus type 2 E3-19K binds to peptide-filled and peptide-deficient HLA-A*1101 molecules. J. Virol. 2005, 79, 13317–13325. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fu, J.; Bouvier, M. Allele- and locus-specific recognition of class I MHC molecules by the immunomodulatory E3-19K protein from adenovirus. J. Immunol. 2007, 178, 4567–4575. [Google Scholar] [CrossRef]
- Fu, J.; Li, L.; Bouvier, M. Adenovirus E3-19K proteins of different serotypes and subgroups have similar, yet distinct, immunomodulatory functions towards major histocompatibility class I molecules. J. Biol. Chem. 2011, 286, 17631–17639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Bouvier, M. Determinants of the endoplasmic reticulum (ER) lumenal-domain of the Adenovirus serotype 2 E3-19K protein for association with and ER-retention of major histocompatibility complex class I molecules. Mol. Immunol. 2011, 48, 532–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flomenberg, P.; Piaskowski, V.; Truitt, R.L.; Casper, J.T. Human adenovirus-specific CD8+ T-cell responses are not inhibited by E3-19K in the presence of gamma interferon. J. Virol. 1996, 70, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.; McMichael, A.; Peterson, P.A. Reduced allorecognition of adenovirus-2 infected cells. J. Immunol. 1987, 138, 3960–3966. [Google Scholar] [PubMed]
- Burgert, H.-G.; Maryanski, J.L.; Kvist, S. “E3/19K” protein of adenovirus type 2 inhibits lysis of cytolytic T lymphocytes by blocking cell surface expression of histocompatibility class I antigens. Proc. Natl. Acad. Sci. USA 1987, 84, 1356–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Tevethia, S.S. Differential effect of adenovirus 2 E3/19K glycoprotein on the expression of H-2Kb and H-2Db class I antigens and H-2Kb- and H-2Db-restricted SV40-specific CTL-mediated lysis. Virology 1988, 165, 357–366. [Google Scholar] [CrossRef]
- Rawle, F.C.; Tollefson, A.E.; Wold, W.S.M.; Gooding, L.R. Mouse anti-adenovirus cytoxic T lymphocytes. J. Immunol. 1989, 138, 3960–3966. [Google Scholar]
- Li, L.; Muzahim, Y.; Bouvier, M. Crystal structure of adenovirus E3-19K bound to HLA-A2 reveals mechanism for immunomodulation. Nat. Struc. Mol. Biol. 2012, 19, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Santarserio, B.D.; Bouvier, M. Structure of the Adenovirus Type 4 (Species E) E3-19K/HLA-A2 complex reveals species-specific features in MHC I recognition. J. Immunol. 2016, 197, 1399–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, E.R.A.; Bouvier, M. Immune evasion by adenoviruses: A window into host-virus adaptation. FEBS Lett. 2019, 593, 3496–3503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.I.; Lee, G.-C.; Chung, J.Y.; Han, T.H.; Lee, Y.K.; Kim, M.S.; Lee, C.H. Detection and molecular characterization of adenoviruses in Korean children hospitalized with acute gastroenteretitis. Microbiol. Immunol. 2012, 56, 523–528. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, G.; Libera, S.D.; Petricca, S.; Iaconelli, M.; Donia, D.; Saccucci, P.; Cenko, F.; Xhelilaj, G.; Divizia, M. Genetic diversity of human adenovírus in children with acute gastreoenteretitis, Albania, 2013–2015. Biomed. Res. Intern. 2015, 2015, 142912. [Google Scholar]
- Afrad, M.H.; Avzun, T.; Haque, J.; Haguw, W.; Hossain, M.E.; Rahman, A.F.M.R.; Ahmed, S.; Faruque, A.S.G.; Rahman, M.Z.; Rahman, M. Detection of enteric- and non-enteric adenoviruses in gastreoenteretitis patients, Bangladesh, 2012–2015. J. Med. Virol. 2017. [Google Scholar] [CrossRef]
- Kumthip, K.; Khamrin, P.; Ushijima, H.; Maneekarn, N. Enteric- and non-enteric adenoviruses associated with acute gastreoenteretitis in pediatric patients in Thailand, 2011 to 2017. PLoS ONE 2019, 14, e0220263. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J.; Telford, A.R.; Watson, M.S.; McBride, K.; Mautner, V. The DNA sequence of adenovirus type 40. J. Mol. Biol. 1993, 234, 1308–1316. [Google Scholar] [CrossRef]
- Bailey, A.; Mautner, V. Phylogenetic relationships among adenovirus serotypes. Virology 1994, 205, 438–452. [Google Scholar] [CrossRef]
- Yeh, H.-Y.; Pieniazek, N.; Pieniazek, D.; Luftig, R. Genetic organization, size, and complete sequence of early region 3 genes of human adenovirus type 41. J. Virol. 1996, 70, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Kosulin, K. Intestinal HAdV infection: Tissue specificity, persistence, and implications for antiviral therapy. Viruses 2019, 11, 804. [Google Scholar] [CrossRef] [Green Version]
- Cepko, C.L.; Whetsone, C.A.; Sharp, P.A. Adenovirus hexon monoclonal antibody that is group specific and potentially useful as a diagnostic reagent. J. Clin. Microbiol. 1983, 17, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotech. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acid Res. 2020, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2011, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.; Jefferys, B. Phyre2: Protein Homology/Analogy Recognition Engine V 2.0; Structural Bioinformatics Group, Imperial College: London, UK, 2011. [Google Scholar]
- Moncaya, G.; Lin, D.; McCarthy, M.T.; Watson, A.A.; O’Calaghan, C.A. MICA expression is regulated by cell adhesion and contact in a FAK/Src-dependent manner. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.S.; Harrach, B.; Ganac, R.D.; Gozum, M.M.A.; de la Cruz, W.P.; Riedel, B.; Pan, C.; Delwart, E.L.; Schnurr, D.P. New adenoviruses species found in a patient presenting with gastroenteritis. J. Virol. 2007, 81, 5978–5984. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Deryckere, F.; Burgert, H.-G. Early region 3 adenovirus type 19 (subgroup D) encodes an HLA-binding protein distinct from that of subgroups B and C. J. Virol. 1996, 70, 2832–2841. [Google Scholar] [CrossRef] [Green Version]
- Rawls-Wilson, J.; Deutscher, S.L.; Wold, W.S.M. The signal-anchor domain of adenovirus E3-6.7K, a type III integral membrane protein can direct adenovirus E3-gp19K, a type I integral membrane protein, into the membrane of the endoplasmic reticulum. Virology 1994, 201, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.A.; Norris, P.S.; Prigozy, T.I.; Bodmer, J.-L.; Mahr, J.A.; Garnett, C.T.; Martinnon, F.; Tschopp, J.; Gooding, L.R.; Ware, C.F. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2. J. Biol. Chem. 2001, 276, 3270–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemessen, C.T.; Kidd, A.H. Adenoviruses type 40 and 41 growth in vitro: Host range diversity reflected by differences in patterns of DNA replication. J. Virol. 1994, 68, 1239–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, J.C. Candidate adenoviruses 40 and 41: Fastidious adenoviruses from human infant stool. J. Med. Virol. 1983, 11, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.J.; Bousquet, E.B. Comparison of enteric adenovirus infection in various human cell lines. J. Virol. Methods 1988, 20, 295–308. [Google Scholar] [CrossRef]
- Kidd, A.H.; Chroboczek, J.; Ruigrok, R.W. Adenovirus type 40 virions contain two distinct fibers. Virology 1983, 192, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.Y.; Pieniazek, N.; Pieniazek, D.; Gelderblom, H.; Luftig, R.B. Human adenovirus type 41 contains two fibers. Virus Res. 1994, 33, 179–198. [Google Scholar] [CrossRef]
- Albinsson, B.; Kidd, A.H. Adenovirus type 41 lacks an RGD alpha(v)-integrin binding motif on the penton base and undergoes delayed uptake in A549 cells. Virus Res. 1999, 64, 125–136. [Google Scholar] [CrossRef]
- Jonjic, S.; Babic, M.; Polic, B.; Krmpotic, A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008, 20, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULPBs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulates NK cytotoxicity through NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Ashiru, O.; Bennett, N.J.; Boyle, L.H.; Thomas, M.; Trowsdale, J.; Wills, M.R. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J. Virol. 2009, 83, 12345–12354. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.L.; Hudson, A.W. The human herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytotoxicity through modulation of MICA and MICB. PLoS Pathog. 2011, 7, e1002362. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Boname, J.M.; Field, S.; Nejentsev, S.; Salio, M.; Cerundolo, V.; Wills, M.; Lehner, P.J. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 1656–1661. [Google Scholar] [CrossRef] [Green Version]
- McSharry, B.P.; Burgert, H.G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sester, M.; Ruszics, Z.; Mackley, E.; Burget, H.-G. Conserved amino acids within the adenovirus 2 E3/19K protein differentially affect downregulation of MHC class I and MICA/B proteins. J. Immunol. 2010, 184, 255–257. [Google Scholar] [CrossRef]
- Heyward, C.Y.; Patel, R.; Mace, E.M.; Grier, J.T.; Guan, H.; Makrygiannis, A.P.; Orange, J.S.; Ricciardi, R.P. Tumorigenic adenovirus 12 cells evade NK cell lysis by reducing the expression of NKG2D ligands. Immunol. Lett. 2012, 4, 16–23. [Google Scholar] [CrossRef]
- Smith, J.G.; Silvestry, M.; Lindert, S.; Lu, W.; Nemerow, G.R.; Stewart, P.L. Insights into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog. 2010, 6, e1000959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holly, M.K.; Smith, J.G. Adenovirus infections of human enteroids reveals interferon sensitivity and preferential infection of globlet cells. J. Virol. 2018, 92, e00250-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, E.R.A.; Li, L.; Bouvier, M. Intracellular Sequestration of the NKG2D Ligand MIC B by Species F Adenovirus. Viruses 2021, 13, 1289. https://doi.org/10.3390/v13071289
Oliveira ERA, Li L, Bouvier M. Intracellular Sequestration of the NKG2D Ligand MIC B by Species F Adenovirus. Viruses. 2021; 13(7):1289. https://doi.org/10.3390/v13071289
Chicago/Turabian StyleOliveira, Edson R. A., Lenong Li, and Marlene Bouvier. 2021. "Intracellular Sequestration of the NKG2D Ligand MIC B by Species F Adenovirus" Viruses 13, no. 7: 1289. https://doi.org/10.3390/v13071289
APA StyleOliveira, E. R. A., Li, L., & Bouvier, M. (2021). Intracellular Sequestration of the NKG2D Ligand MIC B by Species F Adenovirus. Viruses, 13(7), 1289. https://doi.org/10.3390/v13071289