
The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease with Cardiovascular Complications

 , ,
, ,
Abstract
:1. Introduction
2. SARS-CoV-2 and Its Receptors in Cardiac Pathophysiology
2.1. SARS-CoV-2 Receptor Expression and Virion Tropism and Outcome in Heart Tissues
2.2. Atheroprotective Role of ACE2 Receptor, Its Variants and Association with SARS-CoV-2 Susceptibility to Cardiac Risk
3. SARS-CoV-2, and Its Receptors in Blood Vessel Pathophysiology
3.1. Endothelium-Expressed Receptors and Vascular Leakage during SARS-CoV-2 Infection
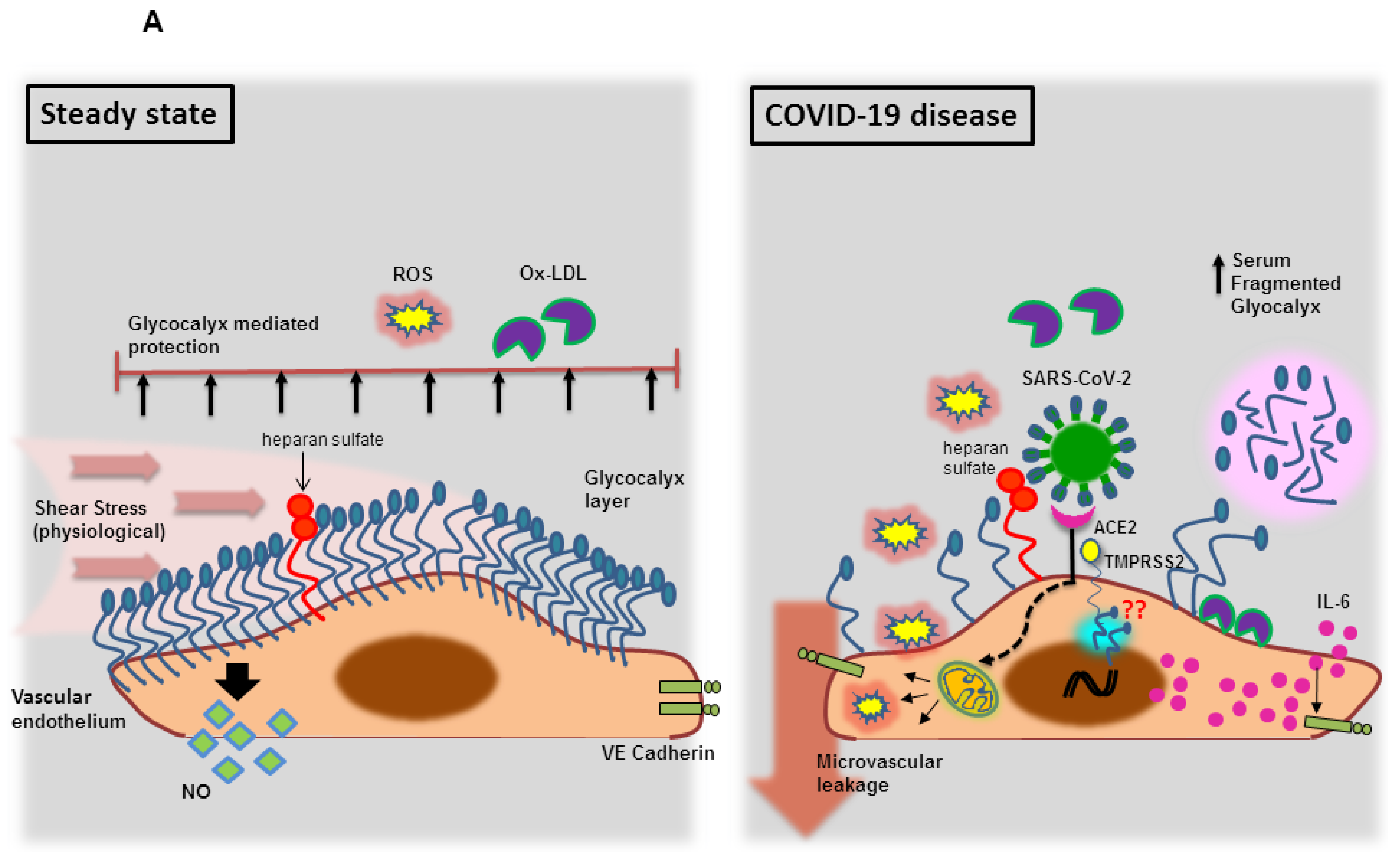
3.1.1. Endothelial Glycocalyx Dysfunction during SARS-CoV-2 Infection
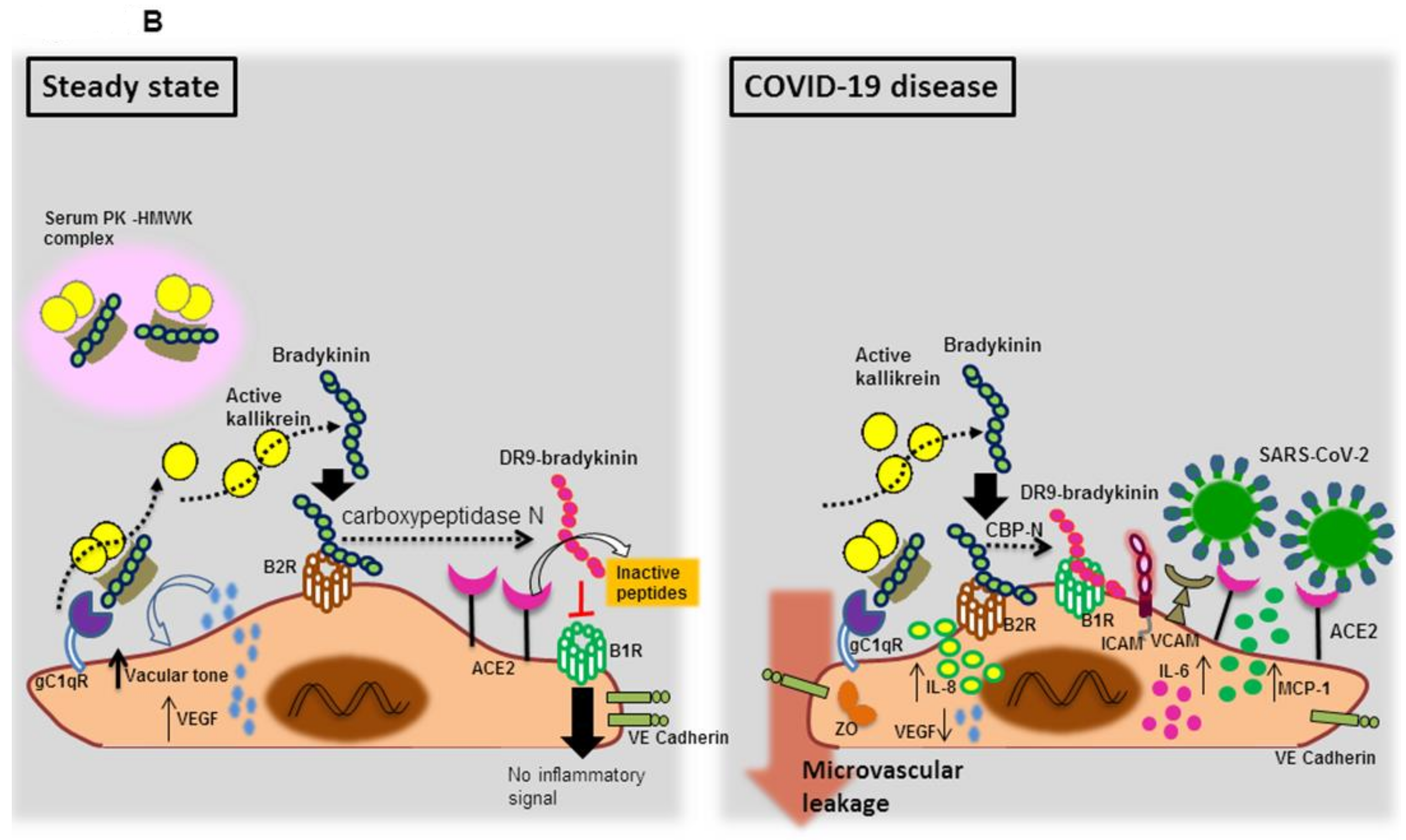
3.1.2. Endothelial Bradykinin 1 Receptor (B1R) Trigger during SARS-CoV-2 Infection
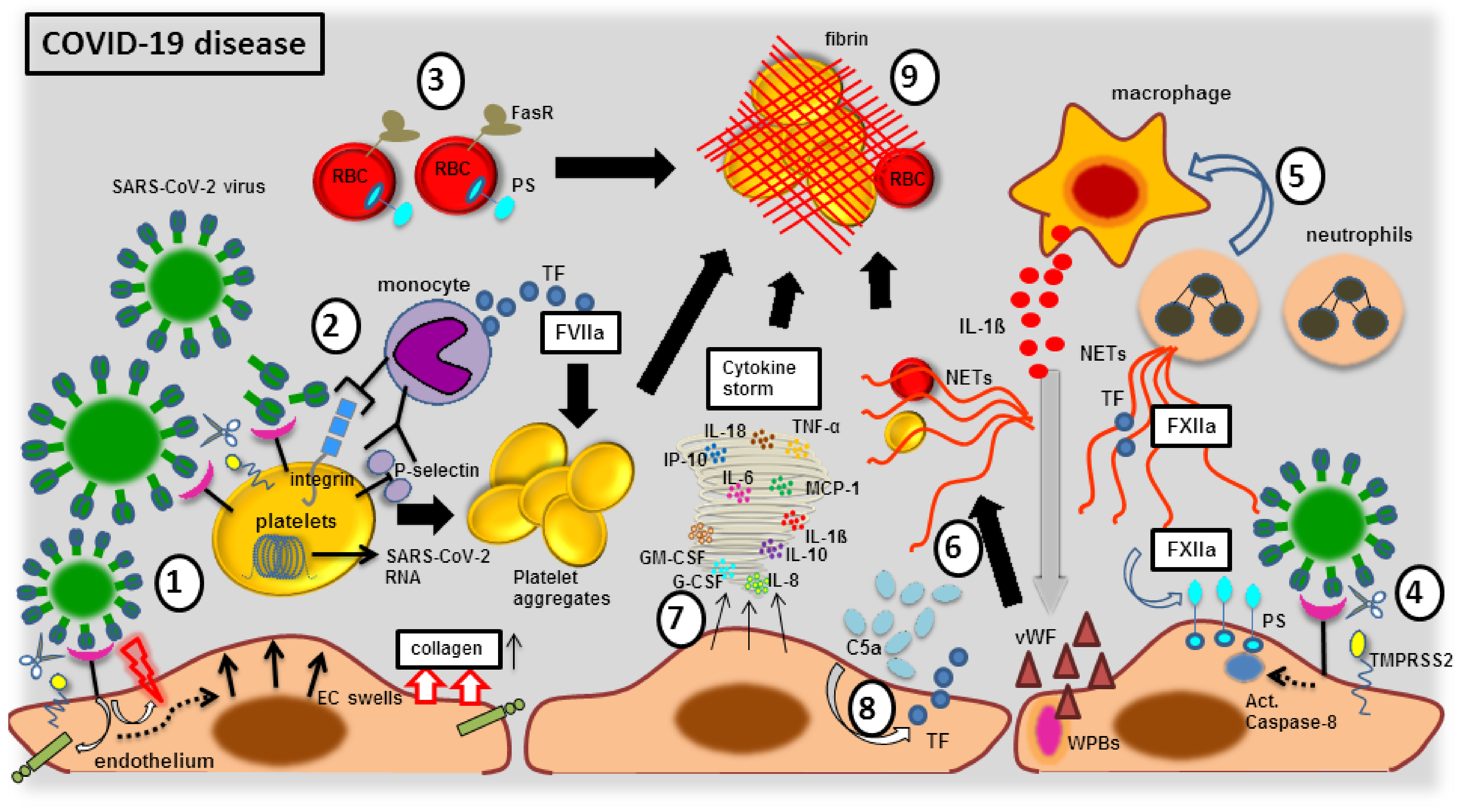
3.2. Endothelial Elemental Changes and Clot Formation during SARS-CoV-2 Infection
3.2.1. The Blood Vessel Clot Formation with FXIIa Factor
3.2.2. The Blood Vessel Clot Formation with Platelet Activation
3.2.3. The Blood Vessel Clot Formation with Complement System Activation
3.2.4. The Blood Vessel Clot Formation with Endothelial Dysfunction
3.2.5. The Blood Vessel Clot Formation with Cellular Immune Response
3.2.6. The Blood Vessel Clot Formation with Cytokine Storm
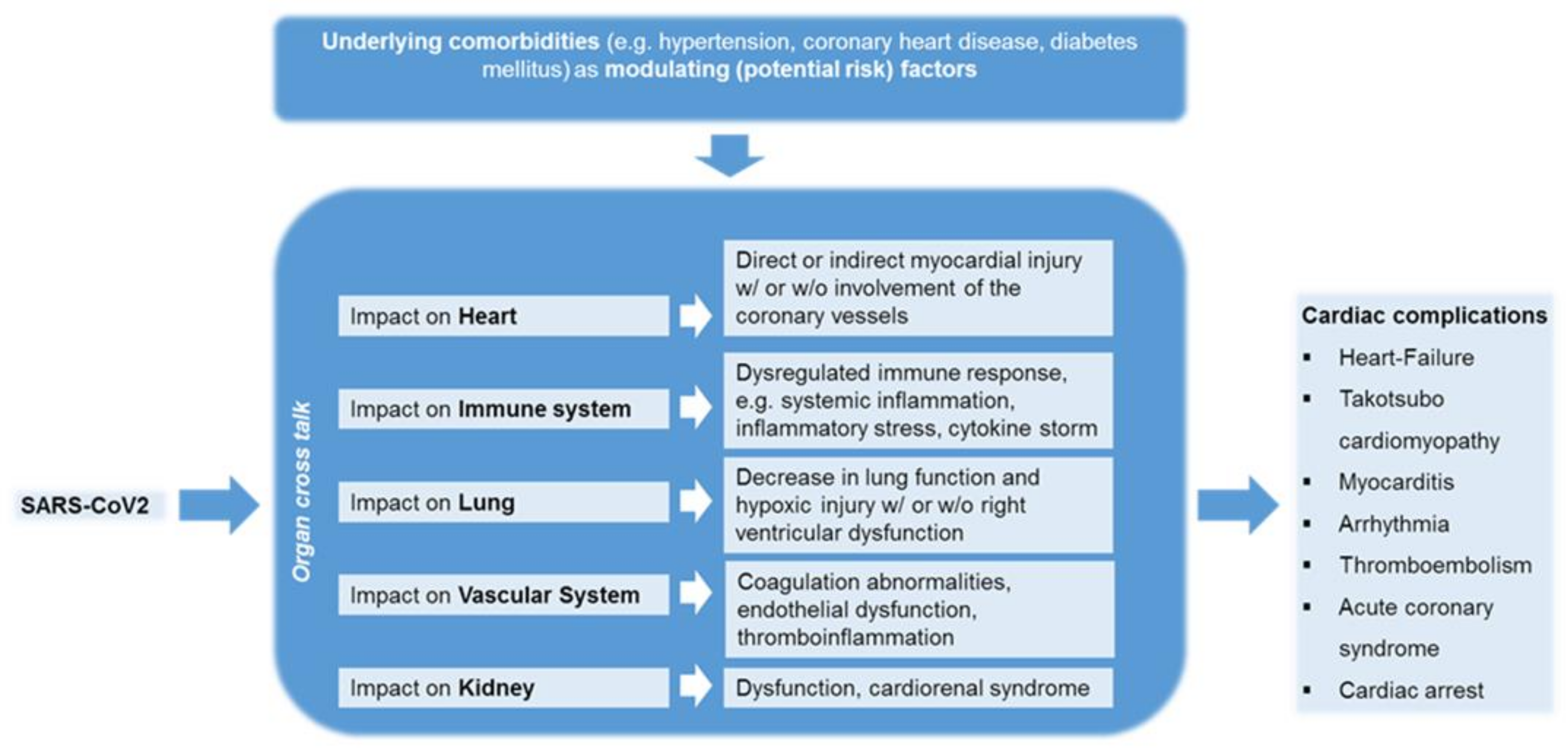
4. Comprehensive Clinical Observations on SARS-CoV-2 Infected Patients Associating with Cardiovascular Complications
4.1. The Impact of COVID-19 on Patients with Cardiovascular Diseases
4.2. The Impact of COVID-19 on Patients without Cardiovascular Diseases
5. Hypothetical Factors Beneath Asymptomatic COVID 19 Patients
6. Different Models to Study the SARS-CoV-2 Virus Interaction with the Cardiovascular System
7. Possible Therapeutic Approaches against SARS-CoV-2 Induced Complications
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus Disease-2019 |
| SARS-CoV-2 | Severe Acute Respiratory Syndr |
| WHO | World Health Organization |
| ACE2 | Angiotensin Converting Enzymes-2 |
| TMPRSS2 | Transmembrane Protease, Serine 2 |
| ECs | Endothelial Cells |
| ROS | Reactive Oxygen Species (ROS) |
| HIF-1α | Hypoxia Inducible Factor-1 alpha |
| STAT-1 | Signal transducer and activator of |
| IL | Interleukin |
| ADAM-17 | Action of a Disintegrin and Metallo |
| TNF-α | Tumor Necrosis Factor-alpha |
| NOX | NADPH oxidase |
| PKR | Protein Kinase R |
| Nrf2 | Nuclear Factor E2-related Factor 2 |
| TLR | Toll-like Receptor |
| ROS | Reactive Oxygen Species |
| VCAM | Vascular Cell Adhesion Molecules |
| TACE | Tumor Necrosis Factor-a Converting Enz |
| RBD | Receptor Binding Domain |
| TGF-β | Transforming Growth Factor-Beta |
| RNA | Ribonucleic Acid |
| RAS | Renin–Angiotensin System |
| CAD | Coronary Artery Disease |
| KKS | Kallikrein–Kinin System |
| B1R and B2R | Bradykinin Receptors |
| DR9-bradykinin | des-Arg-9-bradykinin |
| gC1qR | Globular C1q Receptor |
| Ang II | Angiotensin II |
| ICAM-1 | Intracellular Adhesion Molecules-1 |
| HMWK | High Molecular Weight Kininogen |
| NO | Nitric Oxide |
| eNOS | endothelial nitric oxide synthase |
| vWF | von Willebrand factor |
| NETs | Neutrophil Extracellular Traps |
| TF | Tissue Factor |
| LDL | Low Density Lipoproteins |
| PAI-1 | Plasminogen Activator Inhibitor-1 |
| WPBs | Weibel-Palade bodies |
| ZO | Zonula Occludens |
| DIC | Disseminated Intravascular Coagulation |
| PS | phosphatidylserine |
| RBCs | Red Blood Cells |
| EPCR | Endothelial Cell Protein C Receptor |
| NLRP3 | NLRP- NLR family pyrin domain contain |
| IP-10 | Interferon gamma-induced protein 10 |
| CS | contact system |
| FasR | FS-7-associated surface antigen Recept |
| iPSCs | Induced Pluripotent Stem Cell |
| VTE | Venous Thromboembolism |
| MCP-1 | MCP-1-monocyte chemoattractant prote |
| OHCA | Out-of-Hospital Cardiac Arrest |
| IHCA | In-of-Hospital Cardiac Arrest |
| VEGF | Vascular endothelial Growth Factor |
| IFN-γ | Interferon-gamma |
| GM-CSF | Granulocyte-Monocyte Colony Stimulating Fac |
References
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. Atenei Parm. 2020, 91, 157–160. [Google Scholar]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef]
- Zhong, M.; Lin, B.; Pathak, J.L.; Gao, H.; Young, A.J.; Wang, X.; Liu, C.; Wu, K.; Liu, M.; Chen, J.M.; et al. ACE2 and Furin Expressions in Oral Epithelial Cells Possibly Facilitate COVID-19 Infection via Respiratory and Fecal-Oral Routes. Front. Med. 2020, 7, 580796. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Velavan, T.P.; Meyer, C.G. Mild versus severe COVID-19: Laboratory markers. Int. J. Infect. Dis. 2020, 95, 304–307. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Liu, H.; Gai, S.; Wang, X.; Zeng, J.; Sun, C.; Zhao, Y.; Zheng, Z. Single-cell analysis of SARS-CoV-2 receptor ACE2 and spike protein priming expression of proteases in the human heart. Cardiovasc. Res. 2020, 116, 1733–1741. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, A.; Kawakami, R.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Kutys, R.; Guo, L.; Mori, M.; Cornelissen, A.; Sato, Y.; et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Gkogkou, E.; Barnasas, G.; Vougas, K.; Trougakos, I.P. Expression profiling meta-analysis of ACE2 and TMPRSS2, the putative anti-inflammatory receptor and priming protease of SARS-CoV-2 in human cells, and identification of putative modulators. Redox Biol. 2020, 36, 101615. [Google Scholar] [CrossRef]
- Batlle, M.; Recarte-Pelz, P.; Roig, E.; Castel, M.A.; Cardona, M.; Farrero, M.; Ortiz, J.T.; Campos, B.; Pulgarín, M.J.; Ramírez, J.; et al. AXL receptor tyrosine kinase is increased in patients with heart failure. Int. J. Cardiol. 2014, 173, 402–409. [Google Scholar] [CrossRef] [Green Version]
- DeBerge, M.; Glinton, K.; Subramanian, M.; Wilsbacher, L.D.; Rothlin, C.V.; Tabas, I.; Thorp, E.B. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Bulfamante, G.P.; Perrucci, G.L.; Falleni, M.; Sommariva, E.; Tosi, D.; Martinelli, C.; Songia, P.; Poggio, P.; Carugo, S.; Pompilio, G. Evidence of SARS-CoV-2 Transcriptional Activity in Cardiomyocytes of COVID-19 Patients without Clinical Signs of Cardiac Involvement. Biomedicines 2020, 8, 626. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Ferreira Ferranti, J.; de Almeida Monteiro, R.A.; Duarte-Neto, A.N.; Soares Gomes-Gouvêa, M.; Viu Degaspare, N.; Figueiredo Delgado, A.; Montanari Fiorita, C.; Nunes Leal, G.; Rodrigues, R.M.; et al. SARS-CoV-2 in cardiac tissue of a child with COVID-19-related multisystem inflammatory syndrome. Lancet. Child Adolesc. Health 2020, 4, 790–794. [Google Scholar] [CrossRef]
- Li, N.; Zhu, L.; Sun, L.; Shao, G. The effects of novel coronavirus (SARS-CoV-2) infection on cardiovascular diseases and cardiopulmonary injuries. Stem Cell Res. 2021, 51, 102168. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, E.A.; Eid, N.; Singh, J. Mechanisms of COVID-19-induced heart failure: A short review. Heart Fail. Rev. 2021, 26, 363–369. [Google Scholar] [CrossRef]
- Sharma, A.; Garcia, G., Jr.; Wang, Y.; Plummer, J.T.; Morizono, K.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep. Med. 2020, 1, 100052. [Google Scholar] [CrossRef]
- Bojkova, D.; Wagner, J.U.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxán, G.; Günther, S.; Pham, M.D.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 2020, 116, 2207–2215. [Google Scholar] [CrossRef]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef] [PubMed]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violi, F.; Pastori, D.; Pignatelli, P.; Cangemi, R. SARS-CoV-2 and myocardial injury: A role for Nox2? Intern. Emerg. Med. 2020, 15, 755–758. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Damiano, S.; Sozio, C.; La Rosa, G.; Santillo, M. NOX-Dependent Signaling Dysregulation in Severe COVID-19: Clues to Effective Treatments. Front. Cell. Infect. Microbiol. 2020, 10, 608435. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. TEM 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Gue, Y.X.; Gorog, D.A. Reduction in ACE2 may mediate the prothrombotic phenotype in COVID-19. Eur. Heart J. 2020, 41, 3198–3199. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.H.; Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.T.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xiao, X.; Chen, S.; Zhang, C.; Chen, J.; Yi, D.; Shenoy, V.; Raizada, M.K.; Zhao, B.; Chen, Y. Angiotensin-converting enzyme 2 priming enhances the function of endothelial progenitor cells and their therapeutic efficacy. Hypertension 2013, 61, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes, E.S.A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1–7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Gao, L.; Lu, J.; Zhang, Y.D. ACE2-Ang-(1-7)-Mas Axis in Brain: A Potential Target for Prevention and Treatment of Ischemic Stroke. Curr. Neuropharmacol. 2013, 11, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Stavrou, E.; Schmaier, A.A.; Grobe, N.; Morris, M.; Chen, A.; Nieman, M.T.; Adams, G.N.; LaRusch, G.; Zhou, Y.; et al. Angiotensin 1-7 and Mas decrease thrombosis in Bdkrb2−/− mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 2013, 121, 3023–3032. [Google Scholar] [CrossRef]
- Thatcher, S.E.; Zhang, X.; Howatt, D.A.; Lu, H.; Gurley, S.B.; Daugherty, A.; Cassis, L.A. Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor-/- mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 758–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Li, G.; Guo, J.; Ikezoe, T.; Kasirajan, K.; Zhao, S.; Dalman, R.L. Angiotensin-converting enzyme 2, coronavirus disease 2019, and abdominal aortic aneurysms. J. Vasc. Surg. 2021. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Chiang, A.W.T.; Duong, L.D.; Shoda, T.; Nhu, Q.M.; Ruffner, M.; Hara, T.; Aaron, B.; Joplin, E.; Manresa, M.C.; Abonia, J.P.; et al. Type 2 Immunity and Age Modify Gene Expression of Coronavirus-induced Disease 2019 Receptors in Eosinophilic Gastrointestinal Disorders. J. Pediatric Gastroenterol. Nutr. 2021, 72, 718–722. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Clarke, N.E.; Hooper, N.M.; Turner, A.J. Calmodulin interacts with angiotensin-converting enzyme-2 (ACE2) and inhibits shedding of its ectodomain. FEBS Lett. 2008, 582, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Tang, W.H.; Chen, S.Y.; Van Lente, F.; Francis, G.S.; Sen, S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll. Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Fagyas, M.; Kertész, A.; Siket, I.M.; Bánhegyi, V.; Kracskó, B.; Szegedi, A.; Szokol, M.; Vajda, G.; Rácz, I.; Gulyás, H.; et al. Level of the SARS-CoV-2 receptor ACE2 activity is highly elevated in old-aged patients with aortic stenosis: Implications for ACE2 as a biomarker for the severity of COVID-19. GeroScience 2021, 43, 19–29. [Google Scholar] [CrossRef]
- Ramchand, J.; Patel, S.K.; Kearney, L.G.; Matalanis, G.; Farouque, O.; Srivastava, P.M.; Burrell, L.M. Plasma ACE2 Activity Predicts Mortality in Aortic Stenosis and Is Associated With Severe Myocardial Fibrosis. JACC. Cardiovasc. Imaging 2020, 13, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, J.; Patel, S.K.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef] [PubMed]
- Narula, S.; Yusuf, S.; Chong, M.; Ramasundarahettige, C.; Rangarajan, S.; Bangdiwala, S.I.; van Eikels, M.; Leineweber, K.; Wu, A.; Pigeyre, M.; et al. Plasma ACE2 and risk of death or cardiometabolic diseases: A case-cohort analysis. Lancet 2020, 396, 968–976. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Rodriguez, E.; González-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin II-dependent hypertension. Hypertension 2010, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, T.M.; Al-Sabi, A.; Stockand, J.D. Human recombinant soluble ACE2 (hrsACE2) shows promise for treating severe COVID-19. Signal Transduct. Target. Ther. 2020, 5, 258. [Google Scholar] [CrossRef]
- Wysocki, J.; Schulze, A.; Batlle, D. Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System. Biomolecules 2019, 9, 886. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, J.; Ye, M.; Hassler, L.; Gupta, A.K.; Wang, Y.; Nicoleascu, V.; Randall, G.; Wertheim, J.A.; Batlle, D. A Novel Soluble ACE2 Variant with Prolonged Duration of Action Neutralizes SARS-CoV-2 Infection in Human Kidney Organoids. J. Am. Soc. Nephrol. 2021, 32, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; DeLalio, L.J.; Isakson, B.E.; Wang, T.T. AXL-Mediated Productive Infection of Human Endothelial Cells by Zika Virus. Circ. Res. 2016, 119, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with Covid-19. N. Engl. J. Med. 2021, 384, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Conklin, J.; Frosch, M.P.; Mukerji, S.; Rapalino, O.; Maher, M.; Schaefer, P.W.; Lev, M.H.; Gonzalez, R.G.; Das, S.; Champion, S.N.; et al. Cerebral Microvascular Injury in Severe COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Kim, Y.H.; Nijst, P.; Kiefer, K.; Tang, W.H. Endothelial Glycocalyx as Biomarker for Cardiovascular Diseases: Mechanistic and Clinical Implications. Curr. Heart Fail. Rep. 2017, 14, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Lopatko Fagerström, I.; Ståhl, A.L.; Mossberg, M.; Tati, R.; Kristoffersson, A.C.; Kahn, R.; Bascands, J.L.; Klein, J.; Schanstra, J.P.; Segelmark, M.; et al. Blockade of the kallikrein-kinin system reduces endothelial complement activation in vascular inflammation. EBioMedicine 2019, 47, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Curry, F.E. Layer upon layer: The functional consequences of disrupting the glycocalyx-endothelial barrier in vivo and in vitro. Cardiovasc. Res. 2017, 113, 559–561. [Google Scholar] [CrossRef]
- Machin, D.R.; Phuong, T.T.; Donato, A.J. The role of the endothelial glycocalyx in advanced age and cardiovascular disease. Curr. Opin. Pharmacol. 2019, 45, 66–71. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M. Endothelial glycocalyx damage as a systemic inflammatory microvascular endotheliopathy in COVID-19. Biomed. J. 2020, 43, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M. Vascular Endothelial Glycocalyx Damage in COVID-19. Int. J. Mol. Sci. 2020, 21, 9712. [Google Scholar] [CrossRef]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Alsaffar, H.; Martino, N.; Garrett, J.P.; Adam, A.P. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am. J. Physiol. Cell Physiol. 2018, 314, C589–C602. [Google Scholar] [CrossRef] [Green Version]
- Potje, S.R.; Paula, T.D.; Paulo, M.; Bendhack, L.M. The Role of Glycocalyx and Caveolae in Vascular Homeostasis and Diseases. Front. Physiol. 2020, 11, 620840. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Hilgenfeldt, U.; Puschner, T.; Riester, U.; Finsterle, J.; Hilgenfeldt, J.; Ritz, E. Low-salt diet downregulates plasma but not tissue kallikrein-kinin system. Am. J. Physiol. 1998, 275, F88–F93. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Bryant, J.W.; Shariat-Madar, Z. Human plasma kallikrein-kinin system: Physiological and biochemical parameters. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, F.; Madar, Z.S.; Figueroa, C.D.; Schmaier, A.H. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood 2002, 99, 3585–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shariat-Madar, Z.; Mahdi, F.; Schmaier, A.H. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood 2004, 103, 4554–4561. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Qiu, Q.; Mahdi, F.; Shariat-Madar, Z.; Røjkjaer, R.; Schmaier, A.H. Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1821–H1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; El-Dahr, S.S. Cross-talk of the renin-angiotensin and kallikrein-kinin systems. Biol. Chem. 2006, 387, 145–150. [Google Scholar] [CrossRef]
- Chen, Z.; Deddish, P.A.; Minshall, R.D.; Becker, R.P.; Erdös, E.G.; Tan, F. Human ACE and bradykinin B2 receptors form a complex at the plasma membrane. FASEB J. 2006, 20, 2261–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Vardon-Bounes, F.; Buléon, M.; Guilbeau-Frugier, C.; Séguelas, M.H.; Conil, J.M.; Girolami, J.P.; Tack, I.; Minville, V. Kinin B1 receptor: A potential therapeutic target in sepsis-induced vascular hyperpermeability. J. Transl. Med. 2020, 18, 174. [Google Scholar] [CrossRef] [Green Version]
- Mugisho, O.O.; Robilliard, L.D.; Nicholson, L.F.B.; Graham, E.S.; O’Carroll, S.J. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients with COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, S.; Coppens, M.; van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Müller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Hanify, J.M.; Dupree, L.H.; Johnson, D.W.; Ferreira, J.A. Failure of chemical thromboprophylaxis in critically ill medical and surgical patients with sepsis. J. Crit. Care 2017, 37, 206–210. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Olivé, I.; Sintes, H.; Radua, J.; Abad Capa, J.; Rosell, A. D-dimer in patients infected with COVID-19 and suspected pulmonary embolism. Respir. Med. 2020, 169, 106023. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Gershom, E.S.; Sutherland, M.R.; Lollar, P.; Pryzdial, E.L. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J. Thromb. Haemost. 2010, 8, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Meini, S.; Zanichelli, A.; Sbrojavacca, R.; Iuri, F.; Roberts, A.T.; Suffritti, C.; Tascini, C. Understanding the Pathophysiology of COVID-19: Could the Contact System Be the Key? Front. Immunol. 2020, 11, 2014. [Google Scholar] [CrossRef]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naudin, C.; Burillo, E.; Blankenberg, S.; Butler, L.; Renné, T. Factor XII Contact Activation. Semin. Thromb. Hemost. 2017, 43, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Bednar, V.; Blount, A.; Hogue, B.G. Identification of functionally important negatively charged residues in the carboxy end of mouse hepatitis coronavirus A59 nucleocapsid protein. J. Virol. 2006, 80, 4344–4355. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Ning, B.; Yang, H.S.; Youngquist, B.M.; Niu, A.; Lyon, C.J.; Beddingfield, B.J.; Fears, A.C.; Monk, C.H.; Murrell, A.E.; et al. Sensitive tracking of circulating viral RNA through all stages of SARS-CoV-2 infection. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Englert, H.; Rangaswamy, C.; Deppermann, C.; Sperhake, J.-P.; Krisp, C.; Schreier, D.; Gordon, E.; Konrath, S.; Haddad, M.; Pula, G.; et al. Defective NETs Clearance contributes to sustained FXII Activation in COVID-19-associated Pulmonary Thrombo-Inflammation. bioRxiv 2020, arXiv:2020.12.29.424644. [Google Scholar]
- Jin, X.; Duan, Y.; Bao, T.; Gu, J.; Chen, Y.; Li, Y.; Mao, S.; Chen, Y.; Xie, W. The values of coagulation function in COVID-19 patients. PLoS ONE 2020, 15, e0241329. [Google Scholar] [CrossRef]
- Blasi, A.; von Meijenfeldt, F.A.; Adelmeijer, J.; Calvo, A.; Ibañez, C.; Perdomo, J.; Reverter, J.C.; Lisman, T. In vitro hypercoagulability and ongoing in vivo activation of coagulation and fibrinolysis in COVID-19 patients on anticoagulation. J. Thromb. Haemost. 2020, 18, 2646–2653. [Google Scholar] [CrossRef]
- Tillman, B.; Gailani, D. Inhibition of Factors XI and XII for Prevention of Thrombosis Induced by Artificial Surfaces. Semin. Thromb. Hemost. 2018, 44, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Thon, J.N.; Italiano, J.E. Platelet formation. Semin. Hematol. 2010, 47, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S.; Kolodziej, M.A. Disorders of platelet function. Dis. Mon. 1992, 38, 577–631. [Google Scholar] [CrossRef]
- Tao, L.; Zeng, Q.; Li, J.; Xu, M.; Wang, J.; Pan, Y.; Wang, H.; Tao, Q.; Chen, Y.; Peng, J.; et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J. Hematol. Oncol. 2017, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Senhaji, N.; Darif, Y.; Kojok, K.; Oudghiri, M.; Naya, A. Distinctive roles of PKC delta isozyme in platelet function. Curr. Res. Transl. Med. 2016, 64, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, W.; Guo, Y.; Chen, L.; Zhang, L.; Zhao, S.; Long, D.; Yu, L. Association between platelet parameters and mortality in coronavirus disease 2019: Retrospective cohort study. Platelets 2020, 31, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Handtke, S.; Thiele, T. Large and small platelets-(When) do they differ? J. Thromb. Haemost. 2020, 18, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.C.; Rand, M.L. Procoagulant Phosphatidylserine-Exposing Platelets in vitro and in vivo. Front. Cardiovasc. Med. 2020, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Green, D. Coagulation cascade. Hemodial. Int. 2006, 10 (Suppl.2), S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Ghebrehiwet, B.; Silverberg, M.; Kaplan, A.P. Activation of the classical pathway of complement by Hageman factor fragment. J. Exp. Med. 1981, 153, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gál, P.; Sim, R.B. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Bumiller-Bini, V.; de Freitas Oliveira-Toré, C.; Carvalho, T.M.; Kretzschmar, G.C.; Gonçalves, L.B.; Alencar, N.M.; Gasparetto Filho, M.A.; Beltrame, M.H.; Winter Boldt, A.B. MASPs at the crossroad between the complement and the coagulation cascades—The case for COVID-19. Genet. Mol. Biol. 2021, 44, e20200199. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; ten Cate, H.; van der Poll, T. Endothelium: Interface between coagulation and inflammation. Crit. Care Med. 2002, 30, S220–S224. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Schouten, M.; Wiersinga, W.J.; Levi, M.; van der Poll, T. Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol. 2008, 83, 536–545. [Google Scholar] [CrossRef]
- Giordo, R.; Paliogiannis, P.; Mangoni, A.A.; Pintus, G. SARS-CoV-2 and endothelial cell interaction in COVID-19: Molecular perspectives. Vasc. Biol. 2021, 3, R15–R23. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Wagner, D.D.; Bonfanti, R. von Willebrand factor and the endothelium. Mayo Clin. Proc. 1991, 66, 621–627. [Google Scholar] [CrossRef]
- Ward, S.E.; Curley, G.F.; Lavin, M.; Fogarty, H.; Karampini, E.; McEvoy, N.L.; Clarke, J.; Boylan, M.; Alalqam, R.; Worrall, A.P.; et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): Evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021, 192, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Stockschlaeder, M.; Schneppenheim, R.; Budde, U. Update on von Willebrand factor multimers: Focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul. Fibrinolysis 2014, 25, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Davies, P.F.; Remuzzi, A.; Gordon, E.J.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 2114–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.; Shi, D.; Gordon, M.; Chavda, Y.; Grimaldi, C.; Bajaj, T. Where There’s Smoke, There’s Fire: A Case Report of Turbulent Blood Flow in Lower Extremity Point-of-care Ultrasound in COVID-19. Clin. Pract. Cases Emerg. Med. 2021, 5, 30–34. [Google Scholar]
- Kamel, M.H.; Yin, W.; Zavaro, C.; Francis, J.M.; Chitalia, V.C. Hyperthrombotic Milieu in COVID-19 Patients. Cells 2020, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Cañas, C.A.; Cañas, F.; Bautista-Vargas, M.; Bonilla-Abadía, F. Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin. Appl. Thromb. Hemost. 2021, 27, 10760296211003983. [Google Scholar] [CrossRef] [PubMed]
- McVey, J.H. Tissue factor pathway. Baillieres Clin. Haematol. 1994, 7, 469–484. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell. Mol. Immunol. 2021, 18, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Abassi, Z.; Knaney, Y.; Karram, T.; Heyman, S.N. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 2020, 11, 1312. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.-J.; Yoo, J.-S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–746. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4. [Google Scholar] [CrossRef]
- Pawlinski, R.; Wang, J.G.; Owens, A.P., 3rd; Williams, J.; Antoniak, S.; Tencati, M.; Luther, T.; Rowley, J.W.; Low, E.N.; Weyrich, A.S.; et al. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood 2010, 116, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Lillicrap, D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J. Thromb. Haemost. 2020, 18, 786–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donia, A.; Bokhari, H. Apoptosis induced by SARS-CoV-2: Can we target it? Apoptosis 2021, 26, 7–8. [Google Scholar] [CrossRef]
- Yang, A.; Chen, F.; He, C.; Zhou, J.; Lu, Y.; Dai, J.; Birge, R.B.; Wu, Y. The Procoagulant Activity of Apoptotic Cells Is Mediated by Interaction with Factor XII. Front. Immunol. 2017, 8, 1188. [Google Scholar] [CrossRef] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Goette, A.; Patscheke, M.; Henschke, F.; Hammwohner, M. COVID-19-Induced Cytokine Release Syndrome Associated with Pulmonary Vein Thromboses, Atrial Cardiomyopathy, and Arterial Intima Inflammation. TH Open 2020, 4, e271–e279. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thålin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 988–994. [Google Scholar] [CrossRef]
- Janiuk, K.; Jabłońska, E.; Garley, M. Significance of NETs Formation in COVID-19. Cells 2021, 10, 151. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar]
- Praetner, M.; Zuchtriegel, G.; Holzer, M.; Uhl, B.; Schaubächer, J.; Mittmann, L.; Fabritius, M.; Fürst, R.; Zahler, S.; Funken, D.; et al. Plasminogen Activator Inhibitor-1 Promotes Neutrophil Infiltration and Tissue Injury on Ischemia-Reperfusion. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 622473. [Google Scholar] [CrossRef]
- Whelihan, M.F.; Mann, K.G. The role of the red cell membrane in thrombin generation. Thromb. Res. 2013, 131, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. The red blood cell death receptor and thrombosis. J. Clin. Investig. 2018, 128, 3747–3749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setty, B.N.; Rao, A.K.; Stuart, M.J. Thrombophilia in sickle cell disease: The red cell connection. Blood 2001, 98, 3228–3233. [Google Scholar] [CrossRef] [Green Version]
- Horne, M.K., 3rd; Cullinane, A.M.; Merryman, P.K.; Hoddeson, E.K. The effect of red blood cells on thrombin generation. Br. J. Haematol. 2006, 133, 403–408. [Google Scholar] [CrossRef]
- Sackner, M.A.; Adams, J.A. Endothelial pulsatile shear stress is a backstop for COVID-19. Emerg. Top. Life Sci. 2020, 4, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef] [PubMed]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet. Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Castelli, V.; Cimini, A.; Ferri, C. Cytokine Storm in COVID-19: “When You Come Out of the Storm, You Won’t Be the Same Person Who Walked in”. Front. Immunol. 2020, 11, 2132. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Goyal, A.; Prasad, R.; Goel, P.; Pal, A.; Prasad, S.; Rani, I. An Integrated Approach of the Potential Underlying Molecular Mechanistic Paradigms of SARS-CoV-2-Mediated Coagulopathy. Indian J. Clin. Biochem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF-α and complement component 3 on coagulation. Sci. Rep. 2018, 8, 1812. [Google Scholar] [CrossRef] [Green Version]
- Bester, J.; Matshailwe, C.; Pretorius, E. Simultaneous presence of hypercoagulation and increased clot lysis time due to IL-1β, IL-6 and IL-8. Cytokine 2018, 110, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Della-Torre, E.; Lanzillotta, M.; Campochiaro, C.; Cavalli, G.; De Luca, G.; Tomelleri, A.; Boffini, N.; De Lorenzo, R.; Ruggeri, A.; Rovere-Querini, P.; et al. Respiratory Impairment Predicts Response to IL-1 and IL-6 Blockade in COVID-19 Patients With Severe Pneumonia and Hyper-Inflammation. Front. Immunol. 2021, 12, 675678. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kariwa, H.; Yokota, S.; Iki, S.; Indoh, T.; Yokosawa, N.; Takashima, I.; Tsutsumi, H.; Fujii, N. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 2006, 78, 417–424. [Google Scholar] [CrossRef]
- Yoshida, K.; Taga, T.; Saito, M.; Suematsu, S.; Kumanogoh, A.; Tanaka, T.; Fujiwara, H.; Hirata, M.; Yamagami, T.; Nakahata, T.; et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc. Natl. Acad. Sci. USA 1996, 93, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Liu, B.; Sun, Q.; Zhao, J.; Qi, H.; Li, Q. Platelet factor 4 inhibits IL-17/Stat3 pathway via upregulation of SOCS3 expression in melanoma. Inflammation 2014, 37, 1744–1750. [Google Scholar] [CrossRef]
- Mosnier, L.O. Platelet factor 4 inhibits thrombomodulin-dependent activation of thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin. J. Biol. Chem. 2011, 286, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Martino, N.; Ramos, R.B.; Lu, S.; Leyden, K.; Tomaszek, L.; Jaitovich, A.; Vincent, P.A.; Adam, A.P. Endothelial SOCS3 maintains homeostasis and promotes survival in endotoxemic mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Liu, C.; Su, L.; Zhang, D.; Fan, J.; Yang, Y.; Xiao, M.; Xie, J.; Xu, Y.; et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol. Med. 2020, 26, 97. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Burzynski, L.C.; Humphry, M.; Pyrillou, K.; Wiggins, K.A.; Chan, J.N.E.; Figg, N.; Kitt, L.L.; Summers, C.; Tatham, K.C.; Martin, P.B.; et al. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1α by Thrombin. Immunity 2019, 50, 1033–1042.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Scarpati, E.M.; Sadler, J.E. Regulation of endothelial cell coagulant properties. Modulation of tissue factor, plasminogen activator inhibitors, and thrombomodulin by phorbol 12-myristate 13-acetate and tumor necrosis factor. J. Biol. Chem. 1989, 264, 20705–20713. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Kurosawa, S.; Mollica, J.S.; Ferrell, G.L.; Esmon, C.T. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10212–10216. [Google Scholar] [CrossRef] [Green Version]
- Fukudome, K.; Esmon, C.T. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J. Biol. Chem. 1994, 269, 26486–26491. [Google Scholar] [CrossRef]
- Raadsen, M.; Du Toit, J.; Langerak, T.; van Bussel, B.; van Gorp, E.; Goeijenbier, M. Thrombocytopenia in Virus Infections. J. Clin. Med. 2021, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef]
- van der Poll, T.; Levi, M.; Hack, C.E.; ten Cate, H.; van Deventer, S.J.; Eerenberg, A.J.; de Groot, E.R.; Jansen, J.; Gallati, H.; Büller, H.R.; et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J. Exp. Med. 1994, 179, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2021, 51, 313–329. [Google Scholar] [CrossRef]
- Osnes, L.T.; Westvik, A.B.; Joø, G.B.; Okkenhaug, C.; Kierulf, P. Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulant activity (PCA) in purified human monocytes by IL-4, IL-10 and IL-13. Cytokine 1996, 8, 822–827. [Google Scholar] [CrossRef]
- Qiao, J.; Wu, X.; Luo, Q.; Wei, G.; Xu, M.; Wu, Y.; Liu, Y.; Li, X.; Zi, J.; Ju, W.; et al. NLRP3 regulates platelet integrin αIIbβ3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica 2018, 103, 1568–1576. [Google Scholar] [CrossRef]
- Nosaka, M.; Ishida, Y.; Kimura, A.; Kuninaka, Y.; Inui, M.; Mukaida, N.; Kondo, T. Absence of IFN-γ accelerates thrombus resolution through enhanced MMP-9 and VEGF expression in mice. J. Clin. Investig. 2011, 121, 2911–2920. [Google Scholar] [CrossRef]
- Todoroki, N.; Watanabe, Y.; Akaike, T.; Katagiri, Y.; Tanoue, K.; Yamazaki, H.; Tsuji, T.; Toyoshima, S.; Osawa, T. Enhancement by IL-1 beta and IFN-gamma of platelet activation: Adhesion to leukocytes via GMP-140/PADGEM protein (CD62). Biochem. Biophys. Res. Commun. 1991, 179, 756–761. [Google Scholar] [CrossRef]
- Takahashi, K.; Uwabe, Y.; Sawasaki, Y.; Kiguchi, T.; Nakamura, H.; Kashiwabara, K.; Yagyu, H.; Matsuoka, T. Increased secretion of urokinase-type plasminogen activator by human lung microvascular endothelial cells. Am. J. Physiol. 1998, 275, L47–L54. [Google Scholar] [CrossRef]
- Page, M.J.; Bester, J.; Pretorius, E. Interleukin-12 and its procoagulant effect on erythrocytes, platelets and fibrin(ogen): The lesser known side of inflammation. Br. J. Haematol. 2018, 180, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhou, R.; Zhao, X.; Liu, R.; Ye, C. Correlation between the expression of IL-18 and deep venous thrombosis. Int. J. Mol. Med. 2018, 42, 2972. [Google Scholar]
- Gedefaw, L.; Ullah, S.; Leung, P.H.M.; Cai, Y.; Yip, S.P.; Huang, C.L. Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells 2021, 10, 916. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qu, D.; Esmon, N.L.; Esmon, C.T. Metalloproteolytic release of endothelial cell protein C receptor. J. Biol. Chem. 2000, 275, 6038–6044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, E.F.; Binder, K.; Grell, M.; Scheurich, P.; Pfizenmaier, K. Both tumor necrosis factor receptors, TNFR60 and TNFR80, are involved in signaling endothelial tissue factor expression by juxtacrine tumor necrosis factor alpha. Blood 1995, 86, 1836–1841. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.B.; Jamieson, A.; Tuffin, D.P. The effect of interleukin-4 on tumour necrosis factor-alpha induced expression of tissue factor and plasminogen activator inhibitor-1 in human umbilical vein endothelial cells. Thromb. Haemost. 1993, 70, 1037–1042. [Google Scholar] [CrossRef]
- Williams, M.A.; White, S.A.; Miller, J.J.; Toner, C.; Withington, S.; Newland, A.C.; Kelsey, S.M. Granulocyte-macrophage colony-stimulating factor induces activation and restores respiratory burst activity in monocytes from septic patients. J. Infect. Dis. 1998, 177, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Bönig, H.; Burdach, S.; Göbel, U.; Nürnberger, W. Growth factors and hemostasis: Differential effects of GM-CSF and G-CSF on coagulation activation--laboratory and clinical evidence. Ann. Hematol. 2001, 80, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Schecter, A.D.; Rollins, B.J.; Zhang, Y.J.; Charo, I.F.; Fallon, J.T.; Rossikhina, M.; Giesen, P.L.; Nemerson, Y.; Taubman, M.B. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J. Biol. Chem. 1997, 272, 28568–28573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naing, A.; Infante, J.R.; Papadopoulos, K.P.; Chan, I.H.; Shen, C.; Ratti, N.P.; Rojo, B.; Autio, K.A.; Wong, D.J.; Patel, M.R.; et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8+ T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 2018, 34, 775–791.e3. [Google Scholar] [CrossRef] [Green Version]
- Naing, A.; Papadopoulos, K.P.; Autio, K.A.; Ott, P.A.; Patel, M.R.; Wong, D.J.; Falchook, G.S.; Pant, S.; Whiteside, M.; Rasco, D.R.; et al. Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 3562–3569. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, H.; Dauphars, D.J.; He, Y.W. A Potential Role of Interleukin 10 in COVID-19 Pathogenesis. Trends Immunol. 2021, 42, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Zhang, S.; Yu, M.; Feng, Y.; Long, Q.; Yang, H.; Li, J.; Wang, M. IL-17A promotes the formation of deep vein thrombosis in a mouse model. Int. Immunopharmacol. 2018, 57, 132–138. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Kurz, D.J.; Eberli, F.R. Cardiovascular aspects of COVID-19. Swiss Med. Wkly. 2020, 150, w20417. [Google Scholar]
- Touch, S.; Assmann, K.E.; Aron-Wisnewsky, J.; Marquet, F.; Rouault, C.; Fradet, M.; Mosbah, H.; Consortium, M.; Isnard, R.; Helft, G.; et al. Mucosal-associated invariant T (MAIT) cells are depleted and prone to apoptosis in cardiometabolic disorders. FASEB J. 2018, 32, 5078–5089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrot, T.; Gorin, J.-B.; Ponzetta, A.; Maleki, K.T.; Kammann, T.; Emgård, J.; Perez-Potti, A.; Sekine, T.; Rivera-Ballesteros, O.; Gredmark-Russ, S.; et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol. 2020, 5, eabe1670. [Google Scholar] [CrossRef]
- Ahmed, F.; Jo, D.-H.; Lee, S.-H. Can Natural Killer Cells Be a Principal Player in Anti-SARS-CoV-2 Immunity? Front. Immunol. 2020, 11, 3246. [Google Scholar] [CrossRef]
- Kusnadi, A.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Meckiff, B.J.; Simon, H.; Pelosi, E.; Seumois, G.; Ay, F.; Vijayanand, P.; et al. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8+ T cells. Sci. Immunol. 2021, 6, eabe4782. [Google Scholar] [CrossRef] [PubMed]
- Gronewold, J.; Engels, M.; van de Velde, S.; Cudjoe, T.K.M.; Duman, E.E.; Jokisch, M.; Kleinschnitz, C.; Lauterbach, K.; Erbel, R.; Jöckel, K.H.; et al. Effects of Life Events and Social Isolation on Stroke and Coronary Heart Disease. Stroke 2021, 52, 735–747. [Google Scholar] [CrossRef]
- Muhammad, D.G.; Abubakar, I.A. COVID-19 lockdown may increase cardiovascular disease risk factors. Egypt. Heart J. 2021, 73, 2. [Google Scholar] [CrossRef]
- Adu-Amankwaah, J.; Mprah, R.; Adekunle, A.O.; Ndzie Noah, M.L.; Adzika, G.K.; Machuki, J.O.; Sun, H. The cardiovascular aspect of COVID-19. Ann. Med. 2021, 53, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef]
- Bansal, A.; Kumar, A.; Patel, D.; Puri, R.; Kalra, A.; Kapadia, S.R.; Reed, G.W. Meta-analysis Comparing Outcomes in Patients With and Without Cardiac Injury and Coronavirus Disease 2019 (COVID 19). Am. J. Cardiol. 2021, 141, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, X.; Su, Y.; Ma, J.; Hong, K. Prevalence and impact of cardiac injury on COVID-19: A systematic review and meta-analysis. Clin. Cardiol. 2021, 44, 276–283. [Google Scholar] [CrossRef]
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.J.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Rudraraju, R.; et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 2021, 184, 2167–2182.e22. [Google Scholar] [CrossRef] [PubMed]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the COVID-19 Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerges Harb, J.; Noureldine, H.A.; Chedid, G.; Eldine, M.N.; Abdallah, D.A.; Chedid, N.F.; Nour-Eldine, W. SARS, MERS and COVID-19: Clinical manifestations and organ-system complications: A mini review. Pathog. Dis. 2020, 78, ftaa033. [Google Scholar] [CrossRef] [PubMed]
- Sultanian, P.; Lundgren, P.; Strömsöe, A.; Aune, S.; Bergström, G.; Hagberg, E.; Hollenberg, J.; Lindqvist, J.; Djärv, T.; Castelheim, A.; et al. Cardiac arrest in COVID-19: Characteristics and outcomes of in- and out-of-hospital cardiac arrest. A report from the Swedish Registry for Cardiopulmonary Resuscitation. Eur. Heart J. 2021, 42, 1094–1106. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Kotecha, T.; Knight, D.S.; Razvi, Y.; Kumar, K.; Vimalesvaran, K.; Thornton, G.; Patel, R.; Chacko, L.; Brown, J.T.; Coyle, C.; et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 2021, 42, 1866–1878. [Google Scholar] [CrossRef]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2021, 33, 155–162. [Google Scholar] [CrossRef]
- Talotta, R. Do COVID-19 RNA-based vaccines put at risk of immune-mediated diseases? In reply to “potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases”. Clin. Immunol. 2021, 224, 108665. [Google Scholar] [CrossRef]
- Su, J.R.; McNeil, M.M.; Welsh, K.J.; Marquez, P.L.; Ng, C.; Yan, M.; Cano, M.V. Myopericarditis after vaccination, Vaccine Adverse Event Reporting System (VAERS), 1990–2018. Vaccine 2021, 39, 839–845. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.-R.; Mottaghi, S.S.; Mehrabi, M.-R.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. 2021, 90, 104773. [Google Scholar] [CrossRef]
- Manning, J.T.; Fink, B. Sex differences in the relationship between digit ratio (2D:4D) and national case fatality rates for COVID-19: A reply to Sahin (2020). Early Hum. Dev. 2020, 148, 105120. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Feng, Z.; Li, P.; Yu, Q. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin. Chim. Acta 2020, 509, 220–223. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.; Klein, J.; Sundaram, M.E.; Liu, F.; Wong, P.; Silva, J.; Mao, T.; Oh, J.E.; Mohanty, S.; Huang, J.; et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat. Med. 2021. [Google Scholar] [CrossRef]
- Lau, S.Y.; Wang, P.; Mok, B.W.; Zhang, A.J.; Chu, H.; Lee, A.C.; Deng, S.; Chen, P.; Chan, K.H.; Song, W.; et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 2020, 9, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, E.P.-C.; Hernández-Huerta, M.T.; Mayoral, L.P.-C.; Matias-Cervantes, C.A.; Mayoral-Andrade, G.; Barrios, L.; Ángel, L.; Pérez-Campos, E. Factors related to asymptomatic or severe COVID-19 infection. Med. Hypotheses 2020, 144, 110296. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Dowling, W.E.; Funnell, S.G.P.; Gsell, P.-S.; Riveros-Balta, A.X.; Albrecht, R.A.; Andersen, H.; Baric, R.S.; Carroll, M.W.; Cavaleri, M.; et al. Animal models for COVID-19. Nature 2020, 586, 509–515. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Wong, C.K.; Luk, H.K.; Lai, W.H.; Lau, Y.M.; Zhang, R.R.; Wong, A.C.; Lo, G.C.; Chan, K.H.; Hung, I.F.; Tse, H.F.; et al. Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Platform to Study SARS-CoV-2 Related Myocardial Injury. Circ. J. 2020, 84, 2027–2031. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, S.Y.; Yoon, J.W.; Kim, D.; Yu, S.; Kim, J.S.; Kim, J.H. Cardiotoxicity induced by the combination therapy of chloroquine and azithromycin in human embryonic stem cell-derived cardiomyocytes. BMB Rep. 2020, 53, 545–550. [Google Scholar] [CrossRef]
- Morgan, J.P. Use of the Ferret in Cardiovascular Research. In Biology and Diseases of the Ferret; Wiley: Hoboken, NJ, USA, 2014; pp. 653–663. [Google Scholar]
- Goineau, S.; Pape, D.; Guillo, P.; Ramée, M.P.; Bellissant, E.; Goineau, S.; Pape, D.; Guillo, P.; Ramée, M.P.; Bellissant, E. Hemodynamic and histomorphometric characteristics of dilated cardiomyopathy of Syrian hamsters (Bio TO-2 strain). Can. J. Physiol. Pharmacol. 2001, 79, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.; Olivier, M.; Spradling-Reeves, K.; Karere, G.M.; Comuzzie, A.G.; VandeBerg, J.L. Nonhuman Primates and Translational Research—Cardiovascular Disease. ILAR J. 2017, 58, 235–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heegaard, P.M.H.; Sturek, M.; Alloosh, M.; Belsham, G.J. Animal Models for COVID-19: More to the Picture Than ACE2, Rodents, Ferrets, and Non-human Primates. A Case for Porcine Respiratory Coronavirus and the Obese Ossabaw Pig. Front. Microbiol. 2020, 11, 573756. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Alhazzani, W.; Møller, M.H.; Arabi, Y.M.; Loeb, M.; Gong, M.N.; Fan, E.; Oczkowski, S.; Levy, M.M.; Derde, L.; Dzierba, A.; et al. Surviving Sepsis Campaign: Guidelines on the management of critically ill adults with Coronavirus Disease 2019 (COVID-19). Intensive Care Med. 2020, 46, 854–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Hernandez, M.; Sotomayor, E.M.; Villagra, A. Targeting Macrophages as a Therapeutic Option in Coronavirus Disease 2019. Front. Pharmacol. 2020, 11, 577571. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Hayat, M.; Aryal, D.; Beane, A.; Schultz, M.J. Respiratory Support in COVID-19 Patients, with a Focus on Resource-Limited Settings. Am. J. Trop. Med. Hyg. 2020, 102, 1191–1197. [Google Scholar] [CrossRef]
- Hajra, A.; Mathai, S.V.; Ball, S.; Bandyopadhyay, D.; Veyseh, M.; Chakraborty, S.; Lavie, C.J.; Aronow, W.S. Management of Thrombotic Complications in COVID-19: An Update. Drugs 2020, 80, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hajizadeh, N.; Moore, E.E.; McIntyre, R.C.; Moore, P.; Veress, L.A.; Yaffe, M.B.; Moore, H.B.; Barrett, C.D. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): A case series. J. Thromb. Haemost. 2020, 18, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.; Janssens, U.; Welte, T.; Weber-Carstens, S.; Marx, G.; Karagiannidis, C. German recommendations for critically ill patients with COVID-19. Med. Klin. Intensivmed. Notf. 2020, 115, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine Name and Type | Primary Functions | Potential Role in SARS-CoV-2 Associated Hypercoagulopathy | References |
|---|---|---|---|
| IL-6, proinflammatory |
|
| [203,204,205] |
| IL-1α, proinflammatory |
|
| [206] |
| IL-1ß, proinflammatory |
|
| [160,207] |
| IFN-γ, proinflammatory |
|
| [205,208,209] |
| IL-2, growth factor |
|
| [205,210] |
| IL-12, differentiation factor |
|
| [211] |
| IL-18, proinflammatory |
|
| [212,213] |
| TNFα, proinflammatory |
|
| [201,214,215,216] |
| GM-CSF, hematopoiesis cytokine, growth factor |
|
| [217,218] |
| MCP-1, proinflammatory |
|
| [219] |
| IL-10, anti-inflammatory |
|
| [220,221,222] |
| IL-17A, proinflammatory |
|
| [223] |
| ACE2-Mediated Direct Damage | Hemodynamic Strain of Right Ventricle | Hypoxia-Induced Myocardial Injury | Cardiac Vascular Damage | Systemic Inflammatory Response Syndrome |
|---|---|---|---|---|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veluswamy, P.; Wacker, M.; Stavridis, D.; Reichel, T.; Schmidt, H.; Scherner, M.; Wippermann, J.; Michels, G. The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease with Cardiovascular Complications. Viruses 2021, 13, 1346. https://doi.org/10.3390/v13071346
Veluswamy P, Wacker M, Stavridis D, Reichel T, Schmidt H, Scherner M, Wippermann J, Michels G. The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease with Cardiovascular Complications. Viruses. 2021; 13(7):1346. https://doi.org/10.3390/v13071346
Chicago/Turabian StyleVeluswamy, Priya, Max Wacker, Dimitrios Stavridis, Thomas Reichel, Hendrik Schmidt, Maximilian Scherner, Jens Wippermann, and Guido Michels. 2021. "The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease with Cardiovascular Complications" Viruses 13, no. 7: 1346. https://doi.org/10.3390/v13071346
APA StyleVeluswamy, P., Wacker, M., Stavridis, D., Reichel, T., Schmidt, H., Scherner, M., Wippermann, J., & Michels, G. (2021). The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease with Cardiovascular Complications. Viruses, 13(7), 1346. https://doi.org/10.3390/v13071346