
Oncolytic HSV: Underpinnings of Tumor Susceptibility


Abstract
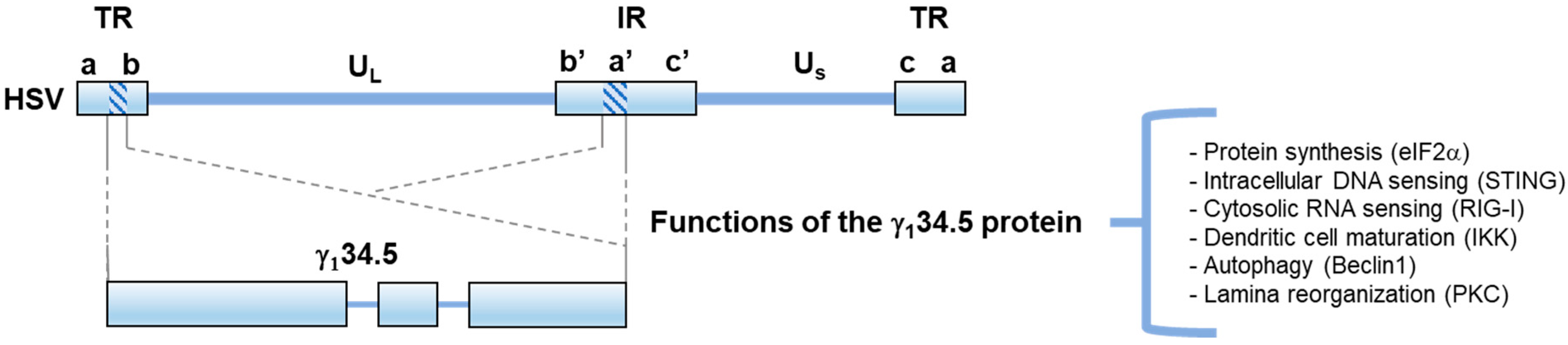
:1. Introduction
2. Selectively Engineered HSV as an Oncolytic Virus
3. HSV Grapples with the Host Cytokine Response
4. Tumor Susceptibility to Δγ134.5 oHSV Replication
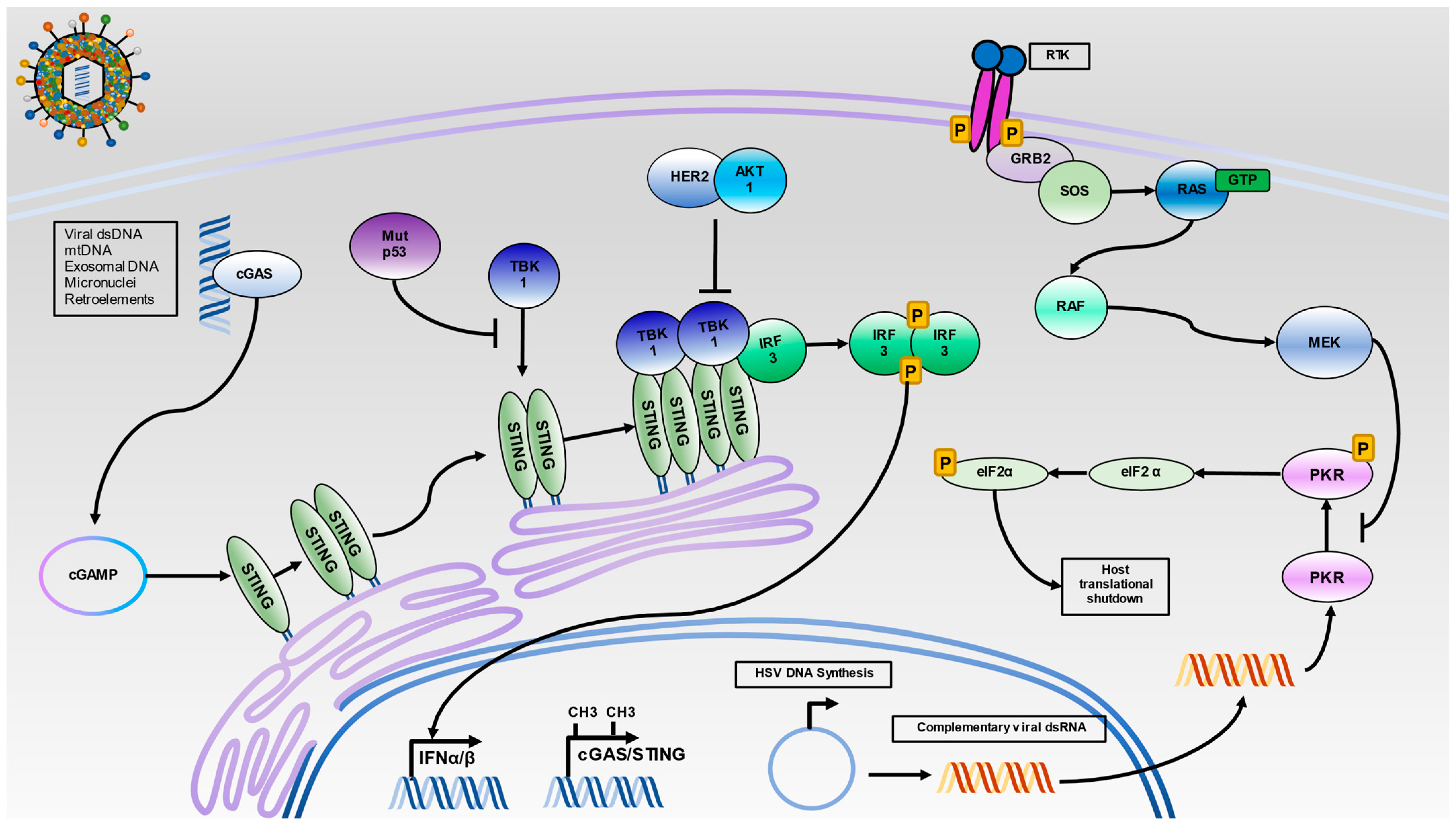
4.1. PKR Pathway Regulation
4.2. STING Pathway Regulation
5. Improving Tumor Selective Replication

{kind=link}
{kind=link}
| Virus | Modifications | Clinical Trials | PMID | Reference |
|---|---|---|---|---|
| Seprehvir (HSV1716) | Deletion of two copies of γ134.5, HSV-1 strain 17 | NCT00931931, NCT01721018, NCT02031965 | 1848598 | [9] |
| NV1020 (R7020) | Deletion of the HSV IR region (deletion of UL56, one copy of ICP0, ICP4, and γ134.5), insertion of US2-2, US2-3, US2-4, and US2-5 derived from HSV-2 | NCT00149396, NCT00012155 | 2842408 | [93] |
| G207 | Deletion of two copies of γ134.5, insertion of lacZ into ICP6 | NCT04482933, NCT03911388, NCT02457845, NCT00028158, NCT00157703 | 7585221 | [94] |
| RP1 | Deletion two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GALV fusogenic protein; insertion of GM-CSF | NCT04050436, NCT03767348, NCT04349436 | 31399043 | [95] |
| RP2 | Deletion of two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GALV fusogenic protein, insertion of anti-CTLA4 antibody | NCT04336241 | 31399043 | [95] |
| RP3 | Deletion of two copies of γ134.5, deletion of ICP47, insertion of GALV fusogenic protein, insertion of proprietary stimulatory agents | NCT04735978 | 33072862 | [96] |
| M032 | Deletion of two copies of γ134.5, expression of IL-12 | NCT02062827 | 27314913 | [97] |
| VG161 | Deletion of two copies of γ134.5; insertion of IL-12, IL-15, IL-15RA, and TF-Fc peptide capable of disrupting PD-1/PD-L1 interactions | NCT04758897, NCT04806464 | 33182232 | [98] |
| ONCR-177 | Deletion of the IR region, null ICP47 mutation; mutations in gB and UL37; insertion of miR-124-3p; insertion of miR-T cassettes targeting HSV-1 genes ICP4, ICP27, UL8, and γ134.5; insertion of anti-CTLA4 (ipilimumab), FLT3LG, CCL4, IL12, IL12B, anti-PD-1 | NCT04348916 | 33355229 | [99] |
| talimogene laherparepvec (T-VEC) | Deletion of two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GM-CSF | >50 Trials in clinicaltrials.gov, Select Trials: NCT00402025, NCT00289016, NCT00769704, NCT02263508, NCT02366195 | 12595888 | [100] |
| OH2 | HSV-2 backbone: Deletion of two copies of γ134.5, ICP47 deletion (α expression of Us11), insertion of GM-CSF | NCT04637698, NCT04616443, NCT04386967, NCT03866525, NCT04755543 | 24671154 | [101] |
| G47Δ | Deletion of two copies of γ134.5, deletion of ICP6, deletion of ICP47 (α expression of Us11) | UMIN000002661, UMIN000010463, UMIN000011636, UMIN000034063 | 11353831 | [102] |
| C134 | Deletion of two copies of γ134.5, insertion of HCMV IRS1 | NCT03657576 | 15994764 | [103] |
| rQNestin34.5v.2 | Single copy of γ134.5 under the nestin promoter | NCT03152318 | 32373649 | [104] |
5.1. α Expression of Us11
5.2. Chimeric oHSV
5.3. Tumor-Selective Expression of γ134.5
5.4. Partial Deletion of γ134.5
6. Improving Antitumor Immunity
7. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ilkow, C.S.; Swift, S.L.; Bell, J.C.; Diallo, J.-S. From Scourge to Cure: Tumour-Selective Viral Pathogenesis as a New Strategy against Cancer. PLoS Pathog. 2014, 10, e1003836. [Google Scholar] [CrossRef] [Green Version]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Diefenbach, R.J.; Miranda-Saksena, M.; Bosnjak, L.; Kim, M.; Jones, C.; Douglas, M.W. The cycle of human herpes simplex virus infection: Virus transport and immune control. J. Infect. Dis. 2006, 194 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 1991, 72, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Chou, J.; Sarmiento, M.; Lerner, R.A.; Roizman, B. Identification by antibody to a synthetic peptide of a protein specified by a diploid gene located in the terminal repeats of the L component of herpes simplex virus genome. J. Virol. 1986, 58, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3266–3270. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, Y.; Voss, K.; van Gent, M.; Chan, Y.K.; Gack, M.U.; Gale, M., Jr.; He, B. The herpesvirus accessory protein γ134.5 facilitates viral replication by disabling mitochondrial translocation of RIG-I. PLoS Pathog. 2021, 17, e1009446. [Google Scholar] [CrossRef]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Jin, H.; Valyi-Nagy, T.; Cao, Y.; Yan, Z.; He, B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 2012, 86, 2188–2196. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Yan, Z.; Ma, Y.; Cao, Y.; He, B. A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and Ikappa B kinase. J. Virol. 2011, 85, 3397–3407. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.M.; MacLean, A.R.; Aitken, J.D.; Harland, J. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J. Gen. Virol. 1994, 75, 3679–3686. [Google Scholar] [CrossRef]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the γ134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Roizman, B. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn+. J. Virol. 1990, 64, 1014–1020. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Bai, X.-C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Cheng, G.; Brett, M.E.; He, B. Val193 and Phe195 of the γ134.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-alpha/beta. Virology 2001, 290, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallóczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.-L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Tallóczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK Suppresses Activation of PKR and Enables Efficient Replication and in vivo Oncolysis by Δ γ134.5 Mutants of Herpes Simplex Virus 1. J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.; Barber, G. Suppression of STING Signaling through Epigenetic Silencing and Missense Mutation Impedes DNA-Damage Mediated Cytokine Production. Oncogene 2018. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e495. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef]
- Jacquemont, B.; Roizman, B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975, 15, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef] [Green Version]
- Leib, D.A.; Machalek, M.A.; Williams, B.R.; Silverman, R.H.; Virgin, H.W. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6097–6101. [Google Scholar] [CrossRef] [Green Version]
- Verpooten, D.; Feng, Z.; Valyi-Nagy, T.; Ma, Y.; Jin, H.; Yan, Z.; Zhang, C.; Cao, Y.; He, B. Dephosphorylation of eIF2alpha mediated by the γ134.5 protein of herpes simplex virus 1 facilitates viral neuroinvasion. J. Virol. 2009, 83, 12626–12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, D.R.; Muller, W.J.; Longnecker, R. HSV targeting of the host phosphatase PP1α is required for disseminated disease in the neonate and contributes to pathogenesis in the brain. Proc. Natl. Acad. Sci. USA 2015, 112, E6937–E6944. [Google Scholar] [CrossRef] [Green Version]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Clemens, M.J. Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene 2004, 23, 3180–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannon, H.S.; Zou, T.; Kiessling, M.K.; Gao, G.F.; Cai, D.; Choi, P.S.; Ivan, A.P.; Buchumenski, I.; Berger, A.C.; Goldstein, J.T.; et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 2018, 9, 5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Q.; Christianson, T.A.; Koretsky, T.; Carlson, H.; David, L.; Keeble, W.; Faulkner, G.R.; Speckhart, A.; Bagby, G.C. Nucleophosmin Interacts with and Inhibits the Catalytic Function of Eukaryotic Initiation Factor 2 Kinase PKR. J. Biol. Chem. 2003, 278, 41709–41717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farassati, F.; Yang, A.-D.; Lee, P.W.K. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 2001, 3, 745–750. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Sun, C.; Luecke, S.; Bodda, C.; Jønsson, K.L.; Cai, Y.; Zhang, B.-C.; Jensen, S.B.; Nordentoft, I.; Jensen, J.M.; Jakobsen, M.R.; et al. Cellular Requirements for Sensing and Elimination of Incoming HSV-1 DNA and Capsids. J. Interferon Cytok. Res. 2019, 39, 191–204. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e1411. [Google Scholar] [CrossRef] [Green Version]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. USA 2019, 116, 11946–11955. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergun, S.L.; Fernandez, D.; Weiss, T.M.; Li, L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019, 178, 290–301.e210. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodda, C.; Reinert, L.S.; Fruhwürth, S.; Richardo, T.; Sun, C.; Zhang, B.-C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergström, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Lin, Y.; Lin, F.; Yang, M.; Li, J.; Zhang, R.; Huang, Z.; Shen, Q.; Tang, R.; Zheng, C. β-Catenin Is Required for the cGAS/STING Signaling Pathway but Antagonized by the Herpes Simplex Virus 1 US3 Protein. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak, C.A.; Classon, M.; De Carvalho, D.D. Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates with Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [Green Version]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Siemers, N.O.; Holloway, J.L.; Chang, H.; Chasalow, S.D.; Ross-MacDonald, P.B.; Voliva, C.F.; Szustakowski, J.D. Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLoS ONE 2017, 12, e0179726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018, 11, 1171–1187. [Google Scholar] [CrossRef]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Pahuja, K.B.; Nguyen, T.T.; Jaiswal, B.S.; Prabhash, K.; Thaker, T.M.; Senger, K.; Chaudhuri, S.; Kljavin, N.M.; Antony, A.; Phalke, S.; et al. Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell 2018, 34, 792–806.e795. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.M.; Advani, S.J.; Bradley, J.D.; Kataoka, Y.; Vashistha, K.; Yan, S.Y.; Markert, J.M.; Gillespie, G.Y.; Whitley, R.J.; Roizman, B.; et al. The use of a genetically engineered herpes simplex virus (R7020) with ionizing radiation for experimental hepatoma. Gene. Ther. 2002, 9, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Andreansky, S.; Soroceanu, L.; Flotte, E.R.; Chou, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997, 57, 1502–1509. [Google Scholar] [PubMed]
- Bennett, J.J.; Delman, K.A.; Burt, B.M.; Mariotti, A.; Malhotra, S.; Zager, J.; Petrowsky, H.; Mastorides, S.; Federoff, H.; Fong, Y. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene. Ther. 2002, 9, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Poon, A.P.; Johnson, J.; Roizman, B. Differential response of human cells to deletions and stop codons in the gamma(1)34.5 gene of herpes simplex virus. J. Virol. 1994, 68, 8304–8311. [Google Scholar] [CrossRef] [Green Version]
- De Queiroz, N.M.G.P.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2019, 17, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Meignier, B.; Longnecker, R.; Roizman, B. In Vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J. Infect. Dis. 1988, 158, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Grandi, P.; Nigim, F. Updates on Oncolytic Virus Immunotherapy for Cancers. Mol. Ther. Oncolytics 2019, 12, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene. Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Chouljenko, D.V.; Ding, J.; Lee, I.F.; Murad, Y.M.; Bu, X.; Liu, G.; Delwar, Z.; Sun, Y.; Yu, S.; Samudio, I.; et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines 2020, 8, 484. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhang, W.; Ning, Z.; Zhuang, X.; Lu, H.; Liang, J.; Li, J.; Zhang, Y.; Dong, Y.; Zhang, Y.; et al. A Novel Oncolytic Herpes Simplex Virus Type 2 Has Potent Anti-Tumor Activity. PLoS ONE 2014, 9, e93103. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Johnson, P.A.; MacLean, C.; Marsden, H.S.; Dalziel, R.G.; Everett, R.D. The product of gene US11 of herpes simplex virus type 1 is expressed as a true late gene. J. Gen. Virol. 1986, 67, 871–883. [Google Scholar] [CrossRef]
- Cassady, K.A.; Gross, M.; Roizman, B. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 1998, 72, 8620–8626. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Matrenec, R.; Gack, M.U.; He, B. Disassembly of the TRIM23-TBK1 Complex by the Us11 Protein of Herpes Simplex Virus 1 Impairs Autophagy. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein US11 Downmodulates the RLR Signaling Pathway via Direct Interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.; Camarena, V.; Mohr, I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J. Virol. 2004, 78, 10193–10196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, I.; Gluzman, Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996, 15, 4759–4766. [Google Scholar] [CrossRef]
- Mohr, I. Neutralizing innate host defenses to control viral translation in HSV-1 infected cells. Int. Rev. Immunol. 2004, 23, 199–220. [Google Scholar] [CrossRef]
- Hardcastle, J.; Kurozumi, K.; Dmitrieva, N.; Sayers, M.P.; Ahmad, S.; Waterman, P.; Weissleder, R.; Chiocca, E.A.; Kaur, B. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol. Ther. 2010, 18, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A.; Saunders, U.; Shimamura, M. Δγ₁134.5 herpes simplex viruses encoding human cytomegalovirus IRS1 or TRS1 induce interferon regulatory factor 3 phosphorylation and an interferon-stimulated gene response. J. Virol. 2012, 86, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene. Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassady, K.A.; Bauer, D.F.; Roth, J.; Chambers, M.R.; Shoeb, T.; Coleman, J.; Prichard, M.; Gillespie, G.Y.; Markert, J.M. Pre-clinical Assessment of C134, a Chimeric Oncolytic Herpes Simplex Virus, in Mice and Non-human Primates. Mol. Ther. Oncolytics 2017, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999, 73, 7556–7564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, H.; Nguyen, T.; Kasai, K.; Passaro, C.; Ito, H.; Goins, W.F.; Shaikh, I.; Erdelyi, R.; Nishihara, R.; Nakano, I.; et al. Toxicity and Efficacy of a Novel GADD34-expressing Oncolytic HSV-1 for the Treatment of Experimental Glioblastoma. Clin. Cancer Res. 2018, 24, 2574–2584. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Chan, M.-K.; Chun, Y.S.; Hezel, M.; Chou, T.-C.; Rusch, V.W.; Fong, Y. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol. Ther. 2006, 5, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarnagin, W.R.; Zager, J.S.; Hezel, M.; Stanziale, S.F.; Adusumilli, P.S.; Gonen, M.; Ebright, M.I.; Culliford, A.; Gusani, N.J.; Fong, Y. Treatment of cholangiocarcinoma with oncolytic herpes simplex virus combined with external beam radiation therapy. Cancer Gene. Ther. 2006, 13, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.M.; Farkaly, T.; Grzesik, P.; Lee, J.; Denslow, A.; Hewett, J.; Bryant, J.; Behara, P.; Goshert, C.; Wambua, D.; et al. Design of an Interferon-Resistant Oncolytic HSV-1 Incorporating Redundant Safety Modalities for Improved Tolerability. Mol. Ther. Oncolytics 2020, 18, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a Fully Retargeted Herpes Simplex Virus 1 Recombinant Capable of Entering Cells Solely via Human Epidermal Growth Factor Receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and Multi-cytokine-Armed Herpes Virus Is a Potent Cancer Endovaccine for Local and Systemic Anti-tumor Treatment. Mol. Ther. Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, B. Selective Editing of Herpes Simplex Virus 1 Enables Interferon Induction and Viral Replication That Destroy Malignant Cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Cerveny, M.; Yang, K.; He, B. Replication of herpes simplex virus 1 depends on the γ134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J. Virol. 2004, 78, 7653–7666. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Jin, H.; Ma, Y.; Prabhakar, B.S.; Feng, Z.; Valyi-Nagy, T.; Yan, Z.; Verpooten, D.; Zhang, C.; Cao, Y.; He, B. The γ134.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J. Virol. 2009, 83, 4984–4994. [Google Scholar] [CrossRef] [Green Version]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-mediated Oncolysis Shapes the Antitumor Immune Response and Contributes to Therapeutic Efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING Signaling in Melanoma Cells Shapes Antigenicity and Can Promote Antitumor T-cell Activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 2018, 24, 2716–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8 + T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Malvehy, J.; Samoylenko, I.; Schadendorf, D.; Gutzmer, R.; Grob, J.-J.; Sacco, J.J.; Gorski, K.S.; Anderson, A.; Pickett, C.A.; Liu, K.; et al. Talimogene laherparepvec upregulates immune-cell populations in non-injected lesions: Findings from a phase II, multicenter, open-label study in patients with stage IIIB-IVM1c melanoma. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients With Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016, 23, 4169–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kangas, C.; Krawczyk, E.; He, B. Oncolytic HSV: Underpinnings of Tumor Susceptibility. Viruses 2021, 13, 1408. https://doi.org/10.3390/v13071408
Kangas C, Krawczyk E, He B. Oncolytic HSV: Underpinnings of Tumor Susceptibility. Viruses. 2021; 13(7):1408. https://doi.org/10.3390/v13071408
Chicago/Turabian StyleKangas, Chase, Eric Krawczyk, and Bin He. 2021. "Oncolytic HSV: Underpinnings of Tumor Susceptibility" Viruses 13, no. 7: 1408. https://doi.org/10.3390/v13071408
APA StyleKangas, C., Krawczyk, E., & He, B. (2021). Oncolytic HSV: Underpinnings of Tumor Susceptibility. Viruses, 13(7), 1408. https://doi.org/10.3390/v13071408