
Virotherapy in Germany—Recent Activities in Virus Engineering, Preclinical Development, and Clinical Studies

 ,
,  ,
,  , ,
, ,  ,
,  , , , ,
, , , ,  ,
, 
 ,
, 
Abstract
:1. Introduction
2. Recent Preclinical Virotherapy Research Activities in Germany
2.1. Adenovirus Platform
2.2. Arenavirus Platform
2.3. Coxsackievirus Platform
2.4. Herpes Simplex Virus Platform
2.5. Measles Vaccine Virus Platform
2.6. Parvovirus Platform
2.7. Vaccinia Virus Platform
2.8. Vesicular Stomatitis Virus Platform
3. Recent Clinical Virotherapy Research Activities in Germany
3.1. H-1 Parvovirus (H-1PV)
3.2. Measles Viruses
3.3. Vaccinia Virus
3.4. Herpes Virus
3.5. Adenovirus
3.6. Coxsackievirus A21
3.7. Reovirus
3.8. Vesicular Stomatitis Virus
4. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breitbach, C.J.; Lichty, B.D.; Bell, J.C. Oncolytic Viruses: Therapeutics with an Identity Crisis. EBioMedicine 2016, 9, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.; Hazini, A.; Seymour, L.W. Tackling HLA Deficiencies Head on with Oncolytic Viruses. Cancers 2021, 13, 719. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Pikor, L.A.; Bell, J.C.; Diallo, J.S. Oncolytic Viruses: Exploiting Cancer’s Deal with the Devil. Trends Cancer 2015, 1, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. Bio. Drugs Clin. Immunother. Biopharm. Gene. Ther. 2019, 33, 485–501. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gürlevik, E.; Ureche, C.I.; Schumacher, A.; Kühnel, F. Oncolytic viruses as anticancer vaccines. Front. Oncol. 2014, 4, 188. [Google Scholar] [CrossRef] [Green Version]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Dorer, D.E.; Nettelbeck, D.M. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv. Drug Deliv. Rev. 2009, 61, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.J.; Russell, S.J. MicroRNAs and oncolytic viruses. Curr. Opin. Virol. 2015, 13, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Pearl, T.M.; Markert, J.M.; Cassady, K.A.; Ghonime, M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics 2019, 13, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Binz, E.; Lauer, U.M. Chemovirotherapy: Combining chemotherapeutic treatment with oncolytic virotherapy. Oncolytic Virotherapy 2015, 4, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [Green Version]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Pol, G.J.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.H.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Federspiel, M.J.; Peng, K.W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Shayakhmetov, D.M. Oncolytic Viruses for Systemic Administration: Engineering a Whole Different Animal. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Chakradhar, S. Viral vanguard: Designing cancer-killing viruses to chase metastatic tumors. Nat. Med. 2017, 23, 652–655. [Google Scholar] [CrossRef]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Ungerechts, G.; Engeland, C.E.; Buchholz, C.J.; Eberle, J.; Fechner, H.; Geletneky, K.; Holm, P.S.; Kreppel, F.; Kühnel, F.; Lang, K.S.; et al. Virotherapy Research in Germany: From Engineering to Translation. Hum. Gene Ther. 2017, 28, 800–819. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, J.; Liu, J.; Wang, H.; Schiwon, M.; Janz, S.; Schaffarczyk, L.; von der Goltz, L.; Ehrke-Schulz, E.; Dörner, J.; et al. An Engineered Virus Library as a Resource for the Spectrum-wide Exploration of Virus and Vector Diversity. Cell Rep. 2017, 19, 1698–1709. [Google Scholar] [CrossRef] [Green Version]
- Mach, N.; Gao, J.; Schaffarczyk, L.; Janz, S.; Ehrke-Schulz, E.; Dittmar, T.; Ehrhardt, A.; Zhang, W. Spectrum-Wide Exploration of Human Adenoviruses for Breast Cancer Therapy. Cancers 2020, 12, 1403. [Google Scholar] [CrossRef]
- Zhang, W.; Mese, K.; Schellhorn, S.; Bahlmann, N.; Mach, N.; Bunz, O.; Dhingra, A.; Hage, E.; Lafon, M.E.; Wodrich, H.; et al. High-Throughput Cloning and Characterization of Emerging Adenovirus Types 70, 73, 74, and 75. Int. J. Mol. Sci. 2020, 21, 6370. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Gackowski, J.; Schmidt, E.; Kochanek, S. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of adenovirus vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, R.F.; Espenlaub, S.; Hoffmeister, A.; Kron, M.W.; Kreppel, F. Covalent decoration of adenovirus vector capsids with the carbohydrate epitope αGal does not improve vector immunogenicity, but allows to study the in vivo fate of adenovirus immunocomplexes. PLoS ONE 2017, 12, e0176852. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, L.; Frank, L.; Wagner, J.; Andreozzi, P.; Hammer, B.; D’Alicarnasso, M.; Pelliccia, M.; Liu, W.; Chakrabortty, S.; et al. Patchy Amphiphilic Dendrimers Bind Adenovirus and Control Its Host Interactions and in Vivo Distribution. ACS Nano 2019, 13, 8749–8759. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.T.; Wrede, C.; Niemann, J.; Brooks, J.; Schwarzer, D.; Kühnel, F.; Gerardy-Schahn, R. Targeting polysialic acid-abundant cancers using oncolytic adenoviruses with fibers fused to active bacteriophage borne endosialidase. Biomaterials 2018, 158, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Woller, N.; Brooks, J.; Fleischmann-Mundt, B.; Martin, N.T.; Kloos, A.; Knocke, S.; Ernst, A.M.; Manns, M.P.; Kubicka, S.; et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nat. Commun. 2019, 10, 3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutzke, L.; Allmendinger, E.; Hirt, K.; Kochanek, S. Chorioallantoic Membrane Tumor Model for Evaluating Oncolytic Viruses. Hum. Gene Ther. 2020, 31, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.C.; Kemker, I.; Krutzke, L.; Allmendinger, E.; Mandell, D.J.; Sewald, N.; Kochanek, S.; Müller, K.M. EGFR-Binding Peptides: From Computational Design towards Tumor-Targeting of Adeno-Associated Virus Capsids. Int. J. Mol. Sci. 2020, 21, 9535. [Google Scholar] [CrossRef]
- Eberle, J. Countering TRAIL Resistance in Melanoma. Cancers 2019, 11, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarif, Z.; Tolksdorf, B.; Fechner, H.; Eberle, J. Mcl-1 targeting strategies unlock the proapoptotic potential of TRAIL in melanoma cells. Mol. Carcinog. 2020, 59, 1256–1268. [Google Scholar] [CrossRef]
- Tolksdorf, B.; Zarif, S.; Eberle, J.; Hazini, A.; Dieringer, B.; Jönsson, F.; Kreppel, F.; Kurreck, J.; Fechner, H. Silencing of Mcl-1 overcomes resistance of melanoma cells against TRAIL-armed oncolytic adenovirus by enhancement of apoptosis. J. Mol. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Procko, E.; Margineantu, D.; Lee, E.F.; Shen, B.W.; Zelter, A.; Silva, D.A.; Chawla, K.; Herold, M.J.; Garnier, J.M.; et al. Computationally designed high specificity inhibitors delineate the roles of BCL2 family proteins in cancer. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Czolk, R.; Schwarz, N.; Koch, H.; Schötterl, S.; Wuttke, T.V.; Holm, P.S.; Huber, S.M.; Naumann, U. Irradiation enhances the therapeutic effect of the oncolytic adenovirus XVir-N-31 in brain tumor initiating cells. Int. J. Mol. Med. 2019, 44, 1484–1494. [Google Scholar] [CrossRef] [Green Version]
- Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenegger, E.; Koll, F.; Haas, H.; Mantwill, K.; Janssen, K.P.; Laschinger, M.; Gschwend, J.; Steiger, K.; Black, P.C.; Moskalev, I.; et al. The Oncolytic Adenovirus XVir-N-31 as a Novel Therapy in Muscle-Invasive Bladder Cancer. Hum. Gene Ther. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Hazini, A.; Pryshliak, M.; Brückner, V.; Klingel, K.; Sauter, M.; Pinkert, S.; Kurreck, J.; Fechner, H. Heparan Sulfate Binding Coxsackievirus B3 Strain PD: A Novel Avirulent Oncolytic Agent Against Human Colorectal Carcinoma. Hum. Gene Ther. 2018, 29, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, J.; Thomas, N.; Plotzky, C.; Rommelaere, J. Modified Rodent Parvovirus Capable of Propagating and Spreading through Human Gliomas. U.S. Patent 9029117, 12 May 2015. [Google Scholar]
- Ferreira, T.; Kulkarni, A.; Bretscher, C.; Richter, K.; Ehrlich, M.; Marchini, A. Oncolytic H-1 Parvovirus Enters Cancer Cells through Clathrin-Mediated Endocytosis. Viruses 2020, 12, 1199. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Ferreira, T.; Bretscher, C.; Grewenig, A.; El-Andaloussi, N.; Bonifati, S.; Marttila, T.; Palissot, V.; Hossain, J.A.; Azuaje, F.; et al. Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry. Nat. Commun. 2021, 12, 3834. [Google Scholar] [CrossRef]
- Abdullahi, S.; Jäkel, M.; Behrend, S.J.; Steiger, K.; Topping, G.; Krabbe, T.; Colombo, A.; Sandig, V.; Schiergens, T.S.; Thasler, W.E.; et al. A Novel Chimeric Oncolytic Virus Vector for Improved Safety and Efficacy as a Platform for the Treatment of Hepatocellular Carcinoma. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Hanauer, J.R.H.; Koch, V.; Lauer, U.M.; Mühlebach, M.D. High-Affinity DARPin Allows Targeting of MeV to Glioblastoma Multiforme in Combination with Protease Targeting without Loss of Potency. Mol. Ther. Oncolytics 2019, 15, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Hazini, A.; Dieringer, B.; Pryshliak, M.; Knoch, K.P.; Heimann, L.; Tolksdorf, B.; Pappritz, K.; El-Shafeey, M.; Solimena, M.; Beling, A.; et al. miR-375- and miR-1-Regulated Coxsackievirus B3 Has No Pancreas and Heart Toxicity But Strong Antitumor Efficiency in Colorectal Carcinomas. Hum. Gene Ther. 2021, 32, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Pinkert, S.; Pryshliak, M.; Pappritz, K.; Knoch, K.; Hazini, A.; Dieringer, B.; Schaar, K.; Dong, F.; Hinze, L.; Lin, J.; et al. Development of a new mouse model for coxsackievirus-induced myocarditis by attenuating coxsackievirus B3 virulence in the pancreas. Cardiovasc. Res. 2020, 116, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Pryshliak, M.; Hazini, A.; Knoch, K.; Dieringer, B.; Tolksdorf, B.; Solimena, M.; Kurreck, J.; Pinkert, S.; Fechner, H. MiR-375-mediated suppression of engineered coxsackievirus B3 in pancreatic cells. FEBS Lett. 2020, 594, 763–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, M.F.; Baertsch, M.A.; Anker, S.C.; Henkel, L.; Singh, H.M.; Bossow, S.; Engeland, C.E.; Barkley, R.; Hoyler, B.; Albert, J.; et al. Enhanced Control of Oncolytic Measles Virus Using MicroRNA Target Sites. Mol. Ther. Oncolytics 2018, 9, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.M.; Leber, M.F.; Bossow, S.; Engeland, C.E.; Dessila, J.; Grossardt, C.; Zaoui, K.; Bell, J.C.; Jäger, D.; von Kalle, C.; et al. MicroRNA-sensitive Oncolytic Measles Virus for Chemovirotherapy of Pancreatic Cancer. Mol. Ther. Oncolytics 2021. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.; Zaun, G.; Hamdan, T.A.; Lang, J.; Adomati, T.; Schmitz, R.; Friedrich, S.K.; Bergerhausen, M.; Cham, L.B.; Li, F.; et al. Arenavirus Induced CCL5 Expression Causes NK Cell-Mediated Melanoma Regression. Front. Immunol. 2020, 11, 1849. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Sharma, P.; Kasper, S.; Helfrich, I.; Pandyra, A.A.; Gassa, A.; Virchow, I.; Flatz, L.; Brandenburg, T.; Namineni, S.; et al. Spatiotemporally restricted arenavirus replication induces immune surveillance and type I interferon-dependent tumour regression. Nat. Commun. 2017, 8, 14447. [Google Scholar] [CrossRef]
- Rajaraman, S.; Canjuga, D.; Ghosh, M.; Codrea, M.C.; Sieger, R.; Wedekink, F.; Tatagiba, M.; Koch, M.; Lauer, U.M.; Nahnsen, S.; et al. Measles Virus-Based Treatments Trigger a Pro-inflammatory Cascade and a Distinctive Immunopeptidome in Glioblastoma. Mol. Ther. Oncolytics 2019, 12, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Goepfert, K.; Dinsart, C.; Rommelaere, J.; Foerster, F.; Moehler, M. Rational Combination of Parvovirus H1 With CTLA-4 and PD-1 Checkpoint Inhibitors Dampens the Tumor Induced Immune Silencing. Front. Oncol. 2019, 9, 425. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, B.; Klein, J.; Delic, M.; Goepfert, K.; Engel, V.; Geberzahn, L.; Lusky, M.; Erbs, P.; Preville, X.; Moehler, M. Immunogenicity of oncolytic vaccinia viruses JX-GFP and TG6002 in a human melanoma in vitro model: Studying immunogenic cell death, dendritic cell maturation and interaction with cytotoxic T lymphocytes. Onco Targets Ther. 2017, 10, 2389–2401. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, B.; Goepfert, K.; Delic, M.; Galle, P.R.; Moehler, M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. Onco Targets Ther. 2013, 6, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backhaus, P.S.; Veinalde, R.; Hartmann, L.; Dunder, J.E.; Jeworowski, L.M.; Albert, J.; Hoyler, B.; Poth, T.; Jäger, D.; Ungerechts, G.; et al. Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists. Viruses 2019, 11, 914. [Google Scholar] [CrossRef] [Green Version]
- Speck, T.; Heidbuechel, J.P.W.; Veinalde, R.; Jaeger, D.; von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy Against Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veinalde, R.; Grossardt, C.; Hartmann, L.; Bourgeois-Daigneault, M.C.; Bell, J.C.; Jäger, D.; von Kalle, C.; Ungerechts, G.; Engeland, C.E. Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through T cell activation. Oncoimmunology 2017, 6, e1285992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, E.; Kubon, K.D.; Mayer, J.K.M.; Pidelaserra-Martí, G.; Albert, J.; Hoyler, B.; Heidbuechel, J.P.W.; Stephenson, K.B.; Lichty, B.D.; Osen, W.; et al. Measles Vaccines Designed for Enhanced CD8(+) T Cell Activation. Viruses 2020, 12, 242. [Google Scholar] [CrossRef] [Green Version]
- Hutzler, S.; Erbar, S.; Jabulowsky, R.A.; Hanauer, J.R.H.; Schnotz, J.H.; Beissert, T.; Bodmer, B.S.; Eberle, R.; Boller, K.; Klamp, T.; et al. Antigen-specific oncolytic MV-based tumor vaccines through presentation of selected tumor-associated antigens on infected cells or virus-like particles. Sci. Rep. 2017, 7, 16892. [Google Scholar] [CrossRef]
- Leber, M.F.; Hoyler, B.; Prien, S.; Neault, S.; Engeland, C.E.; Forster, J.M.; Bossow, S.; Springfeld, C.; von Kalle, C.; Jager, D.; et al. Sequencing of serially passaged measles virus affirms its genomic stability and reveals a nonrandom distribution of consensus mutations. J. Gen. Virol. 2020, 101, 399–409. [Google Scholar] [CrossRef]
- Maurer, S.; Salih, H.R.; Smirnow, I.; Lauer, U.M.; Berchtold, S. Suicide gene-armed measles vaccine virus for the treatment of AML. Int. J. Oncol. 2019, 55, 347–358. [Google Scholar] [CrossRef]
- Berchtold, S.; Beil, J.; Raff, C.; Smirnow, I.; Schell, M.; D’Alvise, J.; Gross, S.; Lauer, U.M. Assessing and Overcoming Resistance Phenomena against a Genetically Modified Vaccinia Virus in Selected Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 7618. [Google Scholar] [CrossRef]
- May, V.; Berchtold, S.; Berger, A.; Venturelli, S.; Burkard, M.; Leischner, C.; Malek, N.P.; Lauer, U.M. Chemovirotherapy for pancreatic cancer: Gemcitabine plus oncolytic measles vaccine virus. Oncol. Lett. 2019, 18, 5534–5542. [Google Scholar] [CrossRef] [Green Version]
- Marchini, A.; Li, J.; Schroeder, L.; Geletneky, K.; Rommelaere, J. Cancer Therapy with a Parvovirus Combined with a bcl-2 Inhibitor. U.S. Patent 9889169, 13 February 2018. [Google Scholar]
- Kloker, L.D.; Berchtold, S.; Smirnow, I.; Beil, J.; Krieg, A.; Sipos, B.; Lauer, U.M. Oncolytic vaccinia virus GLV-1h68 exhibits profound antitumoral activities in cell lines originating from neuroendocrine neoplasms. BMC Cancer 2020, 20, 628. [Google Scholar] [CrossRef]
- Scheubeck, G.; Berchtold, S.; Smirnow, I.; Schenk, A.; Beil, J.; Lauer, U.M. Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus. Viruses 2019, 11, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.; Berchtold, S.; Schmidt, M.; Beil, J.; Smirnow, I.; Venturelli, S.; Burkard, M.; Handgretinger, R.; Lauer, U.M. Biological treatment of pediatric sarcomas by combined virotherapy and NK cell therapy. BMC Cancer 2019, 19, 1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krabbe, T.; Marek, J.; Groll, T.; Steiger, K.; Schmid, R.M.; Krackhardt, A.M.; Altomonte, J. Adoptive T Cell Therapy Is Complemented by Oncolytic Virotherapy with Fusogenic VSV-NDV in Combination Treatment of Murine Melanoma. Cancers 2021, 13, 1044. [Google Scholar] [CrossRef]
- Kloker, L.D.; Berchtold, S.; Smirnow, I.; Schaller, M.; Fehrenbacher, B.; Krieg, A.; Sipos, B.; Lauer, U.M. The Oncolytic Herpes Simplex Virus Talimogene Laherparepvec Shows Promising Efficacy in Neuroendocrine Cancer Cell Lines. Neuroendocrinology 2019, 109, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Recher, M.; Lang, K.S.; Navarini, A.; Hunziker, L.; Lang, P.A.; Fink, K.; Freigang, S.; Georgiev, P.; Hangartner, L.; Zellweger, R.; et al. Extralymphatic virus sanctuaries as a consequence of potent T-cell activation. Nat. Med. 2007, 13, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M. Lymphocytic choriomeningitis virus and immunology. Curr. Top. Microbiol. Immunol. 2002, 263, 1–5. [Google Scholar] [CrossRef]
- Webb, H.E.; Molomut, N.; Padnos, M.; Wetherley-Mein, G. The treatment of 18 cases of malignant disease with an arenavirus. Clin. Oncol. 1975, 1, 157–169. [Google Scholar]
- Geisler, A.; Hazini, A.; Heimann, L.; Kurreck, J.; Fechner, H. Coxsackievirus B3-Its Potential as an Oncolytic Virus. Viruses 2021, 13, 718. [Google Scholar] [CrossRef]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef]
- Schierer, S.; Hesse, A.; Knippertz, I.; Kaempgen, E.; Baur, A.S.; Schuler, G.; Steinkasserer, A.; Nettelbeck, D.M. Human dendritic cells efficiently phagocytose adenoviral oncolysate but require additional stimulation to mature. Int. J. Cancer 2012, 130, 1682–1694. [Google Scholar] [CrossRef]
- Mühlebach, M.D. Vaccine platform recombinant measles virus. Virus Genes 2017, 53, 733–740. [Google Scholar] [CrossRef]
- Hörner, C.; Schürmann, C.; Auste, A.; Ebenig, A.; Muraleedharan, S.; Dinnon, K.H., 3rd; Scholz, T.; Herrmann, M.; Schnierle, B.S.; Baric, R.S.; et al. A highly immunogenic and effective measles virus-based Th1-biased COVID-19 vaccine. Proc. Natl. Acad. Sci. USA 2020, 117, 32657–32666. [Google Scholar] [CrossRef]
- Gogesch, P.; Schülke, S.; Scheurer, S.; Mühlebach, M.D.; Waibler, Z. Modular MLV-VLPs co-displaying ovalbumin peptides and GM-CSF effectively induce expansion of CD11b(+) APC and antigen-specific T cell responses in vitro. Mol. Immunol. 2018, 101, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and combining oncolytic measles virus for cancer therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef]
- Baertsch, M.A.; Leber, M.F.; Bossow, S.; Singh, M.; Engeland, C.E.; Albert, J.; Grossardt, C.; Jager, D.; von Kalle, C.; Ungerechts, G. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene 2014, 21, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Bossow, S.; Leonard, V.H.; Zaoui, K.; Grossardt, C.; Frenzke, M.; Miest, T.; Sawall, S.; Cattaneo, R.; von Kalle, C.; et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Heidbuechel, J.P.W.; Engeland, C.E. Paramyxoviruses for Tumor-targeted Immunomodulation: Design and Evaluation Ex Vivo. J. Vis. Exp. JoVE 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidelaserra-Martí, G.; Engeland, C.E. Mechanisms of measles virus oncolytic immunotherapy. Cytokine Growth Factor Rev. 2020, 56, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef]
- Heidbuechel, J.P.W.; Engeland, C.E. Oncolytic viruses encoding bispecific T cell engagers: A blueprint for emerging immunovirotherapies. J. Hematol. Oncol. 2021, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Lampe, J.; Weiland, T.; Smirnow, I.; Schleicher, S.; Handgretinger, R.; Kopp, H.G.; Reiser, J.; Stubenrauch, F.; Mayer, N.; et al. Innate immune defense defines susceptibility of sarcoma cells to measles vaccine virus-based oncolysis. J. Virol. 2013, 87, 3484–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, T.; Lampe, J.; Essmann, F.; Venturelli, S.; Berger, A.; Bossow, S.; Berchtold, S.; Schulze-Osthoff, K.; Lauer, U.M.; Bitzer, M. Enhanced killing of therapy-induced senescent tumor cells by oncolytic measles vaccine viruses. Int. J. Cancer 2014, 134, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Mousset, S.; Rommelaere, J. Minute virus of mice inhibits cell transformation by simian virus 40. Nature 1982, 300, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef]
- Moehler, M.; Goepfert, K.; Heinrich, B.; Breitbach, C.J.; Delic, M.; Galle, P.R.; Rommelaere, J. Oncolytic virotherapy as emerging immunotherapeutic modality: Potential of parvovirus h-1. Front. Oncol. 2014, 4, 92. [Google Scholar] [CrossRef] [Green Version]
- Angelova, A.; Ferreira, T.; Bretscher, C.; Rommelaere, J.; Marchini, A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers 2021, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, A.; Kavishwar, G.; Salvato, I.; Marchini, A. A Roadmap for the Success of Oncolytic Parvovirus-Based Anticancer Therapies. Annu. Rev. Virol. 2020, 7, 537–557. [Google Scholar] [CrossRef]
- Hallauer, C.; Kronauer, G.; Siegl, G. Parvoiruses as contaminants of permanent human cell lines. I. Virus isolation from 1960-1970. Arch. Gesamte Virusforsch 1971, 35, 80–90. [Google Scholar] [CrossRef]
- Foloppe, J.; Kempf, J.; Futin, N.; Kintz, J.; Cordier, P.; Pichon, C.; Findeli, A.; Vorburger, F.; Quemeneur, E.; Erbs, P. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Mol. Ther. Oncolytics 2019, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Nasar, F.; Coleman, J.W.; Price, R.E.; Javadian, A.; Draper, K.; Lee, M.; Reilly, P.A.; Clarke, D.K.; Hendry, R.M.; et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 2007, 360, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Krabbe, T.; Altomonte, J. Fusogenic Viruses in Oncolytic Immunotherapy. Cancers 2018, 10, 216. [Google Scholar] [CrossRef] [Green Version]
- Altomonte, J.; Marozin, S.; Schmid, R.M.; Ebert, O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 275–284. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joos, S.; Nettelbeck, D.M.; Reil-Held, A.; Engelmann, K.; Moosmann, A.; Eggert, A.; Hiddemann, W.; Krause, M.; Peters, C.; Schuler, M.; et al. German Cancer Consortium (DKTK)—A national consortium for translational cancer research. Mol. Oncol. 2019, 13, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Bartsch, A.; Weiss, C.; Bernhard, H.; Marchini, A.; Rommelaere, J. ATIM-40. High rate of objective anti-tumor response in 9 patients with glioblastoma after viro-immunotherapy with oncolytic parvovirus H-1 in combination with bevacizumab and PD-1 checkpoint blockade. Neuro-Oncology 2018, 20, vi10. [Google Scholar] [CrossRef] [Green Version]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-based oncolytic immunotherapy Pexastimogene Devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: A randomized multicenter Phase IIb trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-pd-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]


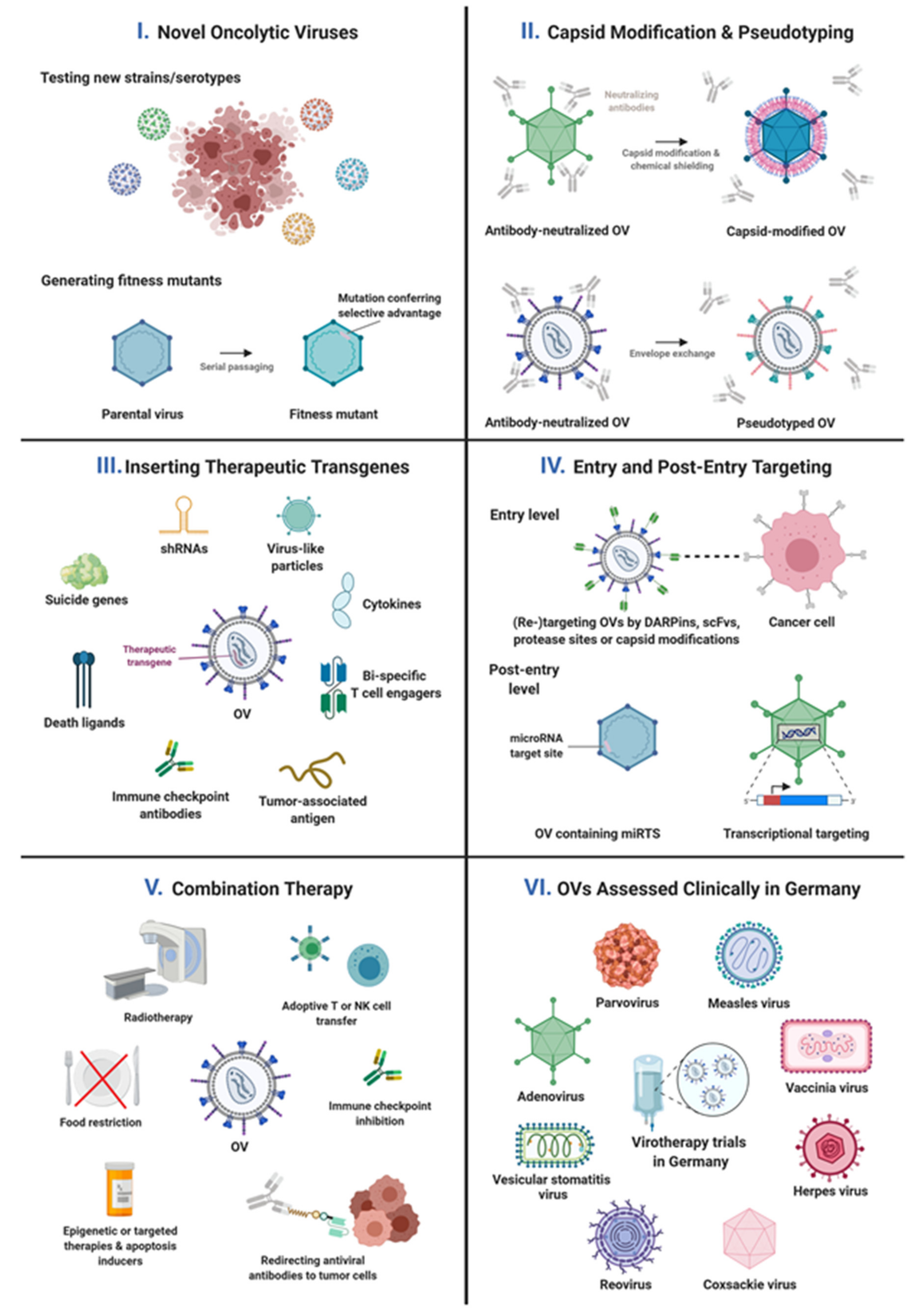{kind=link}
| Scientific Strategy | Description of Research Approach | Virus | Refs |
|---|---|---|---|
| Identifying new viruses as OVs | Screening and/or cloning of virus strains, serotypes, or mutants | adenovirus coxsackievirus parvovirus | [29,30,31] [46] [47] |
| Shielding virus particles from blood factors or from cellular sequestration | Combining genetic and chemical capsid engineering or exploiting adapter molecules for shielding and targeting of virus particles and for exploration of host interactions | adenovirus | [33,34] |
| Exploring carrier cells to enable systemic administration of OVs | adenovirus | on-going work | |
| Exploring/Targeting/Enhancing efficiency of OV cell binding and entry | Unraveling the virus cell entry pathway | parvovirus | [48,49] |
| Genetically replacing the cell-binding domain of a viral capsid protein with a tumor-specific ligand | adenovirus | [35] | |
| Genetic engineering of virus capsid for enhanced entry into tumor cells, cells of the TME, and carrier cells | adenovirus | Nilson et al., submitted, on-going work | |
| Replacing OV glycoproteins by those of other viruses | VSV | [50] | |
| Genetic engineering of viral glycoproteins using highly stable and affine targeting domains and selected protease recognition motifs for combined receptor and protease targeting | measles virus | [51] | |
| Combining genetic and chemical capsid engineering or exploiting adapter molecules for shielding and targeting of virus particles and exploration of host interactions | adenovirus | [33,34] | |
| Post-entry targeting of OV replication | Expression of essential viral genes from tumor-selective promoters | adenovirus | on-going work |
| Insertion of microRNA target sites into viral genes for mRNA destruction and/or translational inhibition in healthy tissues | coxsackievirus measles virus | [52,53,54] [55,56] | |
| Enhancing oncolytic activity or tumor-specificity of OVs | Enhancing oncolytic activity of OVs: production of fitness mutants with enhanced oncosuppressive capacity | coxsackievirus parvovirus | on-going work [47] |
| Enhancing the tumor-specificity of OVs by selecting mutated viruses in a fast evolution platform | arenavirus | on-going work | |
| Immune effects of OVs and enhancing their immuno-stimulatory potency | OV-induced activation of innate and (anti-tumor) adaptive immunity | arenavirus measles virus parvovirus vaccinia virus | [57,58] [59] [60] [61,62] |
| Enabling OV-induced syncytia formation as immunogenic cell death by replacing viral glycoproteins with heterologous fusogenic envelope proteins | VSV | [50] | |
| Expression of immunomodulators (ICIs, bispecifics, cytokines) | adenovirus coxsackievirus measles virus | on-going work on-going work [63,64,65] | |
| Expression (and presentation) of tumor antigens for genetic vaccination | measles virus | [66,67] | |
| OV stability | Analysis of genomic stability of OVs | measles virus | [68] |
| Expression of therapeutic proteins or shRNAs by OVs | Induction of apoptosis by expression of death ligands or RNAi-mediated inhibition of anti-apoptotic proteins of intrinsic apoptosis pathways | adenovirus | [41] and on-going work |
| Insertion of suicide genes into the virus genome for genetic prodrug activation | measles virus vaccinia virus | [51,56,69] [61,70] | |
| Expression of immunomodulators (ICIs, bispecifics, cytokines) | adenovirus coxsackievirus measles virus | on-going work on-going work [63,64,65] | |
| Expression (and presentation) of tumor antigens for genetic vaccination | measles virus | [66,67] | |
| Combination therapy with OVs | Combination therapy with radiotherapy | adenovirus measles virus | [43] on-going work |
| Combination therapy with chemotherapy | measles virus vaccinia virus | [71] [61] | |
| Combination therapy with apoptosis induction | adenovirus parvovirus | [41] and on-going work [72] | |
| Combination with targeted therapy | adenovirus measles virus vaccinia virus | [44] and on-going work on-going work [73] | |
| Combination with epigenetic therapy | adenovirus | on-going work | |
| Combination therapy with starvation | measles virus | [74] | |
| Combination therapy with ICI | adenovirus parvovirus | [36] [60,62] | |
| Combination therapy with adoptive T cell or NK cell transfer | measles virus VSV | [75] [76] | |
| Combination therapy with anti-viral antibody-retargeting via recombinant adapters | adenovirus | [36] | |
| Control of OV replication (safety measure) | OV inhibition by virostatic drugs | herpes virus | [77] |
| Virus Platform | Virus Name | Transgene | Combined With | Name | Identifier | Entity | Phase | Status |
|---|---|---|---|---|---|---|---|---|
| PV | ParvOryx/ H1-PV | ParvOryx01 | Eudra-CT 2011-000572-33 | GBM | I/IIa | C | ||
| ParvOryx/ H1-PV | ParvOryx02 | Eudra-CT 2015-001119-11 | Metastatic PDAC | II | C | |||
| MeV | MeV-IL12 | IL-12 | CanVirex01 | GI basket trial | I/II | P | ||
| MeV-SCD | SCD | 5-FC + ICI | NSCLC | P | ||||
| MeV-SCD | SCD | 5-FC + ICI | GI basket trial | P | ||||
| VV | Pexa-Vec/ JX-594 | GM-CSF | TRAVERSE | NCT01387555 | HCC | IIb | C | |
| Pexa-Vec/ JX-594 | GM-CSF | sorafenib | PHOCUS | NCT02562755 | HCC | III | T | |
| HSV | T-Vec/ Imlygic | GM-CSF | ICI | MASTERKEY-265 | NCT02263508 | Melanoma | Ib/III | T |
| T-Vec/ Imlygic | GM-CSF | ICI | Eudra-CT 2015-005480-16 | TNBC and CRC with liver metastases | Ib | O | ||
| T-Vec/ Imlygic | GM-CSF | ICI | Eudra-CT 2019-001906-61 | Melanoma | II | O | ||
| T-Vec/ Imlygic | GM-CSF | ICI | Eudra-CT 2014-005386-67 | HCC & non-HCC liver metastases | Ib/II | O | ||
| RP-1 | GM-CSF, GALV-GP-R- | ICI | CERPASS | Eudra-CT 2018-003964-30 | CSCC | II | O | |
| AdV | AdVince | CPA | RADNET | Eudra-CT 2014-000614-64 | NET with liver metastases | I/IIa | O | |
| PeptiCRAd-1 | CD40L, OX40L | ICI | START | Eudra-CT 2021-000642-18 | Basket trial | I | P | |
| CoxV | V937/CVA21 | ICI | NCT04521621 | Basket trial | Ib/II | O | ||
| ReoV | Pelareorep | ICI | GOBLET | Eudra-CT 2020-003996-16 | GI basket trial | I/II | P | |
| VSV | VSV-GP | GP of LCMV | ICI | Basket trial | I | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nettelbeck, D.M.; Leber, M.F.; Altomonte, J.; Angelova, A.; Beil, J.; Berchtold, S.; Delic, M.; Eberle, J.; Ehrhardt, A.; Engeland, C.E.; et al. Virotherapy in Germany—Recent Activities in Virus Engineering, Preclinical Development, and Clinical Studies. Viruses 2021, 13, 1420. https://doi.org/10.3390/v13081420
Nettelbeck DM, Leber MF, Altomonte J, Angelova A, Beil J, Berchtold S, Delic M, Eberle J, Ehrhardt A, Engeland CE, et al. Virotherapy in Germany—Recent Activities in Virus Engineering, Preclinical Development, and Clinical Studies. Viruses. 2021; 13(8):1420. https://doi.org/10.3390/v13081420
Chicago/Turabian StyleNettelbeck, Dirk M., Mathias F. Leber, Jennifer Altomonte, Assia Angelova, Julia Beil, Susanne Berchtold, Maike Delic, Jürgen Eberle, Anja Ehrhardt, Christine E. Engeland, and et al. 2021. "Virotherapy in Germany—Recent Activities in Virus Engineering, Preclinical Development, and Clinical Studies" Viruses 13, no. 8: 1420. https://doi.org/10.3390/v13081420
APA StyleNettelbeck, D. M., Leber, M. F., Altomonte, J., Angelova, A., Beil, J., Berchtold, S., Delic, M., Eberle, J., Ehrhardt, A., Engeland, C. E., Fechner, H., Geletneky, K., Goepfert, K., Holm, P. S., Kochanek, S., Kreppel, F., Krutzke, L., Kühnel, F., Lang, K. S., ... Ungerechts, G. (2021). Virotherapy in Germany—Recent Activities in Virus Engineering, Preclinical Development, and Clinical Studies. Viruses, 13(8), 1420. https://doi.org/10.3390/v13081420