
Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Primary Human AML Cells and Xenotransplantation
2.3. Alpharetroviral Vector Constructs
2.4. Alpharetroviral Vector Supernatant Production and Cell Modification
2.5. Immunoblot
2.6. In Vitro Cytotoxicity Assay
2.7. Cytokine Analyses
2.8. Statistical Analyses
3. Results
3.1. Modification of NK-92 Cells with Alpharetroviral CAR Vectors
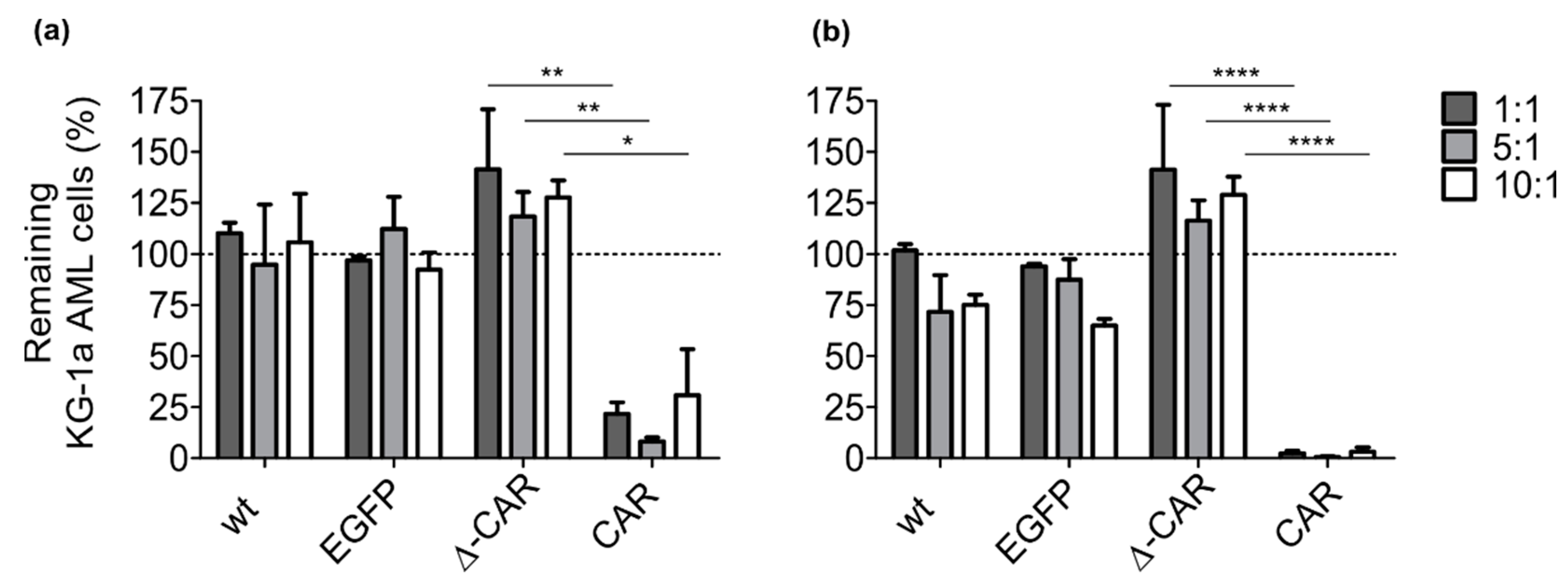
3.2. Improved Anti-AML Activity of CAR-NK-92 Cells In Vitro
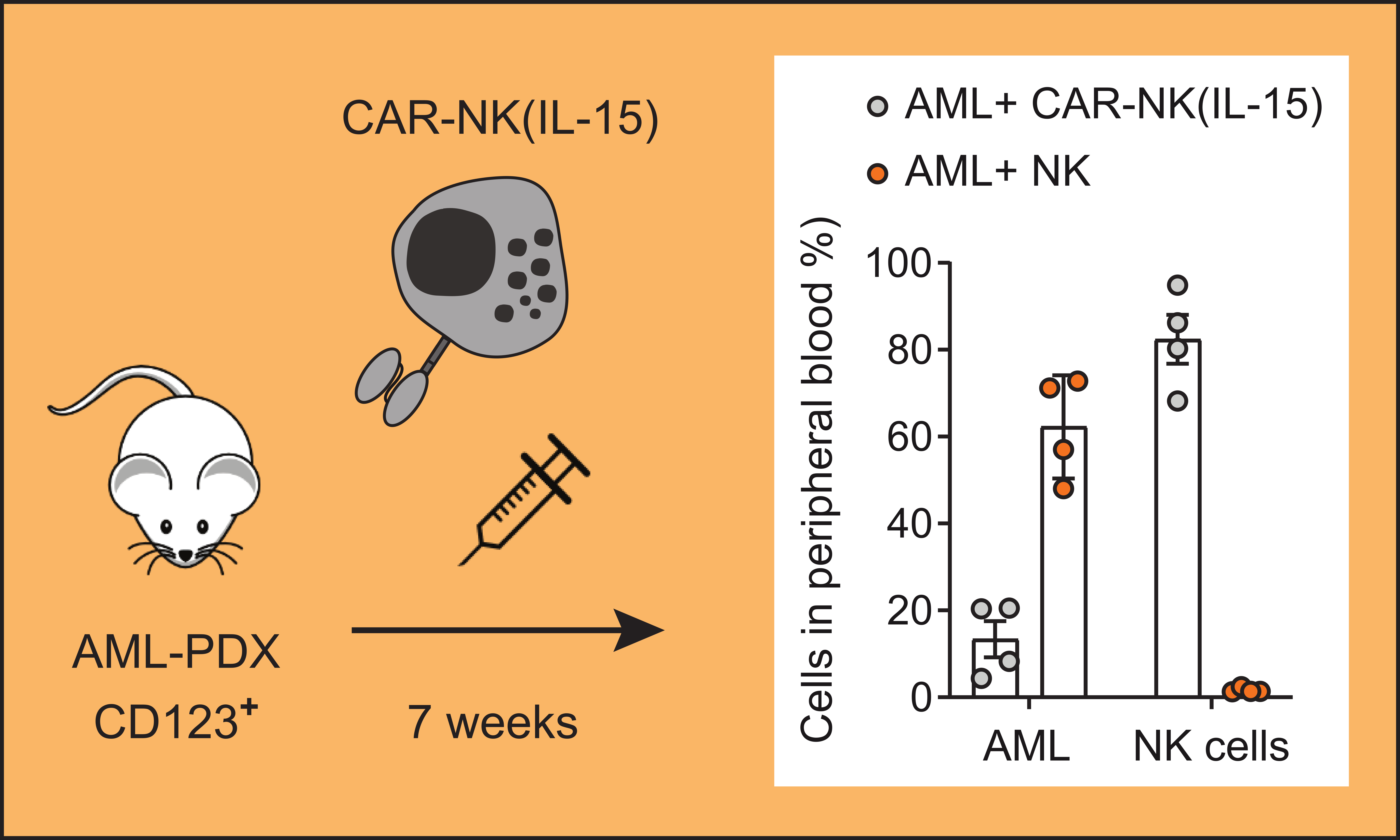
3.3. CAR-NK-92 Cells Exhibit In Vivo Anti-AML Activity in an AML-PDX Model
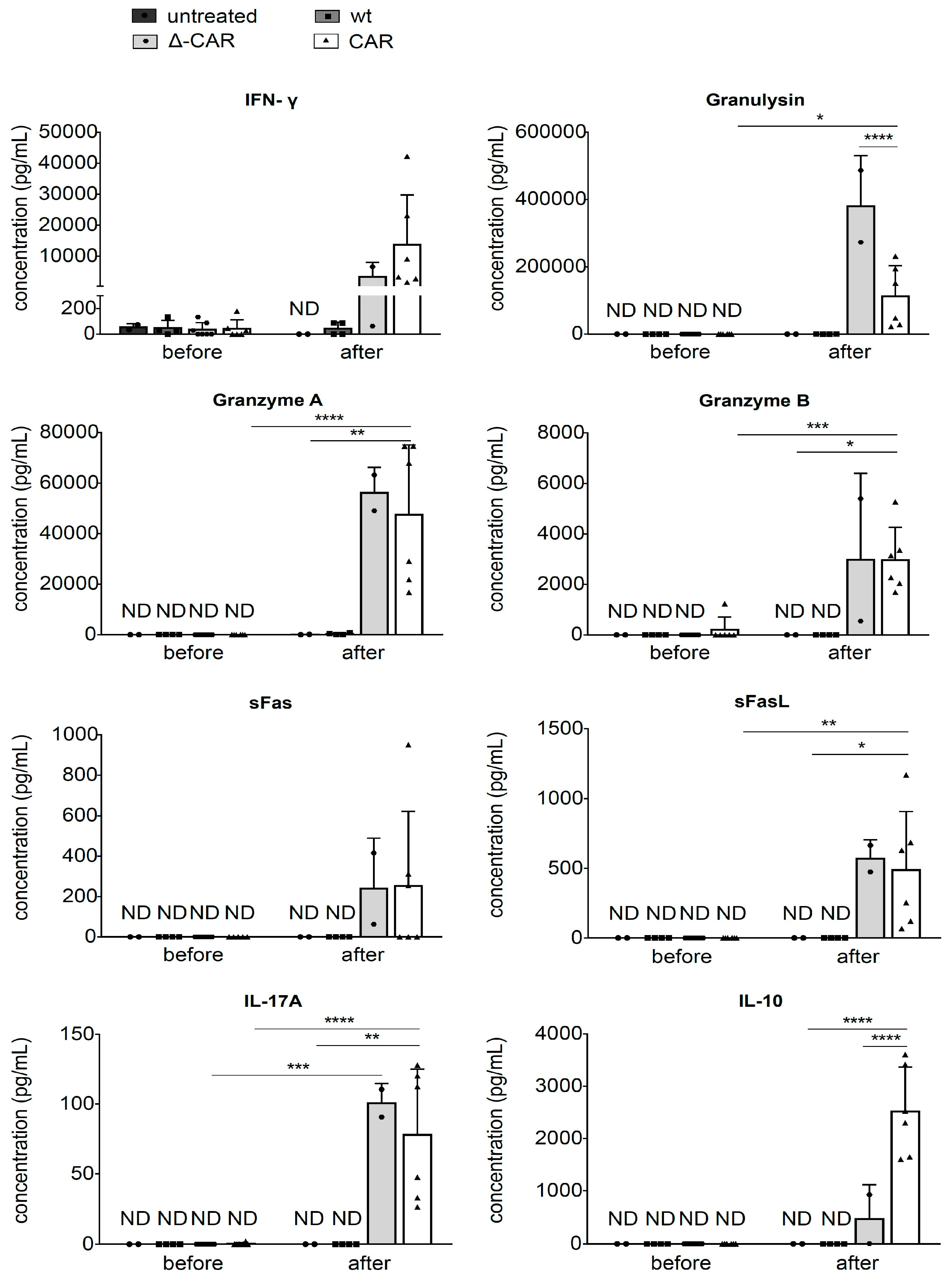
3.4. Secretion of Degranulation Markers In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suerth, J.D.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Moiani, A.; Suerth, J.D.; Gandolfi, F.; Rizzi, E.; Severgnini, M.; De Bellis, G.; Schambach, A.; Mavilio, F. Genome-wide analysis of alpharetroviral integration in human hematopoietic stem/progenitor cells. Genes 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T-cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T-cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T-cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T-cells Are Associated with High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadie, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, A.E.; de Haas, V.; van Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; Te Marvelde, J.G.; Zwaan, C.M.; van Dongen, J.J.M.; Leusen, J.H.W.; van der Velden, V.H.J. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytometry 2018, 96, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Busfield, S.J.; Biondo, M.; Wong, M.; Ramshaw, H.S.; Lee, E.M.; Ghosh, S.; Braley, H.; Panousis, C.; Roberts, A.W.; He, S.Z.; et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 2014, 28, 2213–2221. [Google Scholar] [CrossRef]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.C.; Bretzlaff, W.; et al. T-cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loff, S.; Dietrich, J.; Meyer, J.E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Grunder, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T-cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Kloess, S.; Oberschmidt, O.; Dahlke, J.; Vu, X.K.; Neudoerfl, C.; Kloos, A.; Gardlowski, T.; Matthies, N.; Heuser, M.; Meyer, J.; et al. Preclinical Assessment of Suitable Natural Killer Cell Sources for Chimeric Antigen Receptor Natural Killer-Based “Off-the-Shelf” Acute Myeloid Leukemia Immunotherapies. Hum. Gene Ther. 2019, 30, 381–401. [Google Scholar] [CrossRef]
- Charan, J.; Kantharia, N.D. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013, 4, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Suerth, J.D.; Morgan, M.A.; Kloess, S.; Heckl, D.; Neudorfl, C.; Falk, C.S.; Koehl, U.; Schambach, A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J. Mol. Med. 2016, 94, 83–93. [Google Scholar] [CrossRef]
- Maetzig, T.; Kuehle, J.; Schwarzer, A.; Turan, S.; Rothe, M.; Chaturvedi, A.; Morgan, M.; Ha, T.C.; Heuser, M.; Hammerschmidt, W.; et al. All-in-One inducible lentiviral vector systems based on drug controlled FLP recombinase. Biomaterials 2014, 35, 4345–4356. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Dubois, S.; Conlon, K.C.; Muller, J.R.; Hsu-Albert, J.; Beltran, N.; Bryant, B.R.; Waldmann, T.A. IL15 Infusion of Cancer Patients Expands the Subpopulation of Cytotoxic CD56(bright) NK Cells and Increases NK-Cell Cytokine Release Capabilities. Cancer Immunol. Res. 2017, 5, 929–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, S.H.; Yoon, S.R.; Park, Y.J.; Jung, H.; Kim, T.D.; Choi, I. IL-15-induced IL-10 increases the cytolytic activity of human natural killer cells. Mol. Cells 2011, 32, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Shimizu, K.; Shimizu, T.; Lotze, M.T. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood 2001, 98, 2143–2151. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, J.; Mumm, J.B.; Chan, I.H.; LaFace, D.; Truong, H.; McClanahan, T.; Gorman, D.M.; Oft, M. IL-10 directly activates and expands tumor-resident CD8(+) T-cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012, 72, 3570–3581. [Google Scholar] [CrossRef] [Green Version]
- Littwitz-Salomon, E.; Malyshkina, A.; Schimmer, S.; Dittmer, U. The Cytotoxic Activity of Natural Killer Cells Is Suppressed by IL-10(+) Regulatory T-cells During Acute Retroviral Infection. Front. Immunol. 2018, 9, 1947. [Google Scholar] [CrossRef]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Baessler, T.; Charton, J.E.; Schmiedel, B.J.; Grunebach, F.; Krusch, M.; Wacker, A.; Rammensee, H.G.; Salih, H.R. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood 2010, 115, 3058–3069. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.R.; Clara, J.A.; Reger, R.N.; Allan, D.S.J.; Childs, R.W. RNA-Seq Analysis Reveals CCR5 as a Key Target for CRISPR Gene Editing to Regulate In Vivo NK Cell Trafficking. Cancers 2021, 13, 872. [Google Scholar] [CrossRef]
- Levy, E.; Reger, R.; Segerberg, F.; Lambert, M.; Leijonhufvud, C.; Baumer, Y.; Carlsten, M.; Childs, R. Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4(R334X). Front. Immunol. 2019, 10, 1262. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzel, C.; Sun, Z.; Cripe, L.D.; Fernandez, H.F.; Douer, D.; Rowe, J.M.; Paietta, E.M.; Ketterling, R.; O’Connell, M.J.; Wiernik, P.H.; et al. Very poor long-term survival in past and more recent studies for relapsed AML patients: The ECOG-ACRIN experience. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Willier, S.; Rothamel, P.; Hastreiter, M.; Wilhelm, J.; Stenger, D.; Blaeschke, F.; Rohlfs, M.; Kaeuferle, T.; Schmid, I.; Albert, M.H.; et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood 2021, 137, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J., Jr. High-affinity FRbeta-specific CAR T-cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3-specific Chimeric Antigen Receptor T-cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.A.; Wang, X.H.; Leyton, J.V.; Maghera, S.; Deif, B.; Reilly, R.M.; Minden, M.D.; Keating, A. CD16(+)NK-92 and anti-CD123 monoclonal antibody prolongs survival in primary human acute myeloid leukemia xenografted mice. Haematologica 2018, 103, 1720–1729. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, M.A.; Kloos, A.; Lenz, D.; Kattre, N.; Nowak, J.; Bentele, M.; Keisker, M.; Dahlke, J.; Zimmermann, K.; Sauer, M.; et al. Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123. Viruses 2021, 13, 1365. https://doi.org/10.3390/v13071365
Morgan MA, Kloos A, Lenz D, Kattre N, Nowak J, Bentele M, Keisker M, Dahlke J, Zimmermann K, Sauer M, et al. Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123. Viruses. 2021; 13(7):1365. https://doi.org/10.3390/v13071365
Chicago/Turabian StyleMorgan, Michael A., Arnold Kloos, Daniela Lenz, Nadine Kattre, Juliette Nowak, Marco Bentele, Maximilian Keisker, Julia Dahlke, Katharina Zimmermann, Martin Sauer, and et al. 2021. "Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123" Viruses 13, no. 7: 1365. https://doi.org/10.3390/v13071365
APA StyleMorgan, M. A., Kloos, A., Lenz, D., Kattre, N., Nowak, J., Bentele, M., Keisker, M., Dahlke, J., Zimmermann, K., Sauer, M., Heuser, M., & Schambach, A. (2021). Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123. Viruses, 13(7), 1365. https://doi.org/10.3390/v13071365