
The Influence of Habitat on Viral Diversity in Neotropical Rodent Hosts

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical and Legal Statements
2.2. Capturing
2.3. Sample Processing
2.3.1. Nuclease Treatment and Viral Nucleic Acid Extraction
2.3.2. Reverse Transcription and Amplification
2.4. Next-Generation Sequencing
2.5. Bioinformatic Analyses
2.6. Statistical Analysis
2.7. Phylogenetic Analyses
Nucleotide Sequence Accession Numbers
3. Results
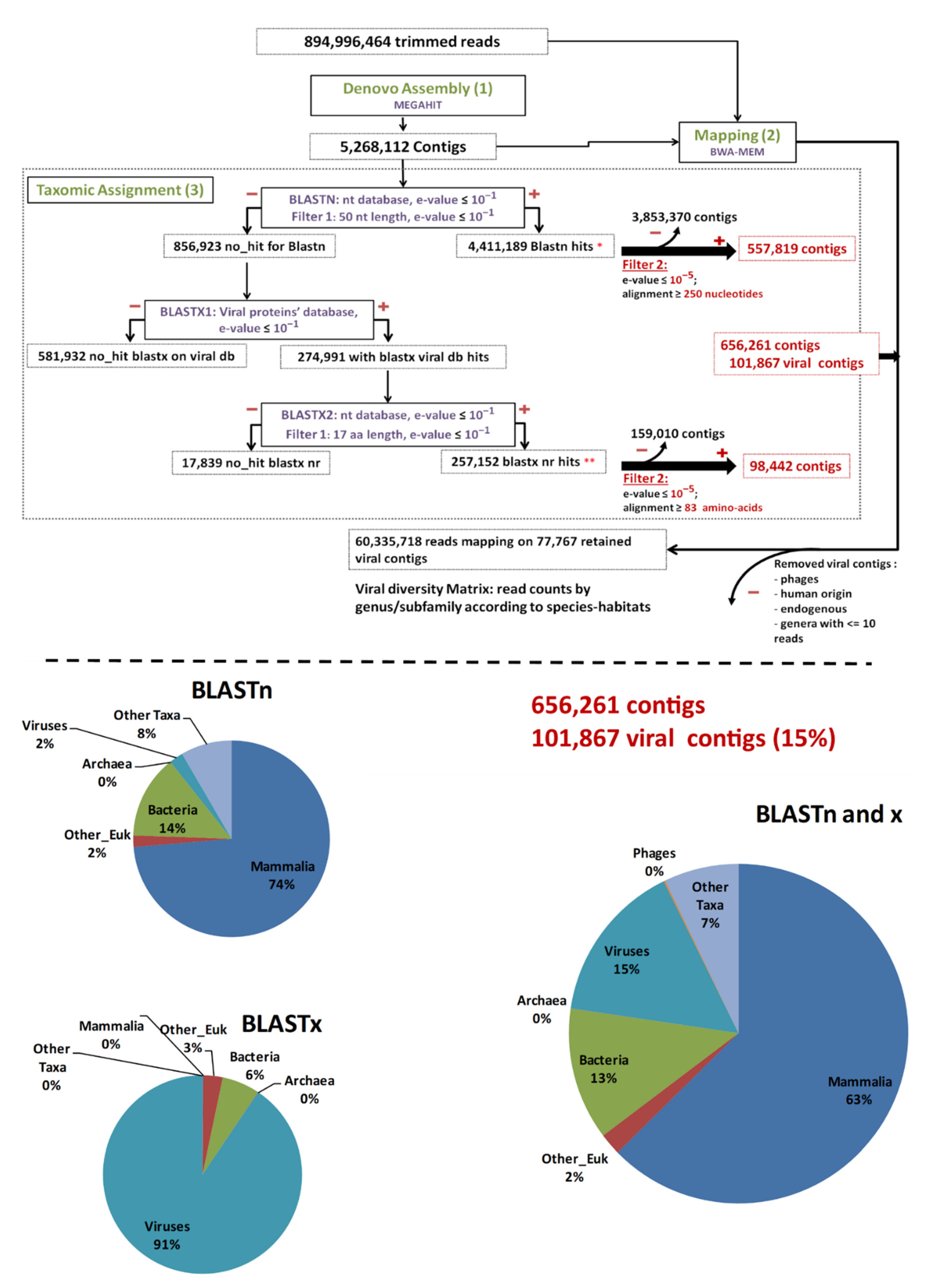
3.1. Illumina Sequencing and Bioinformatics Analyses
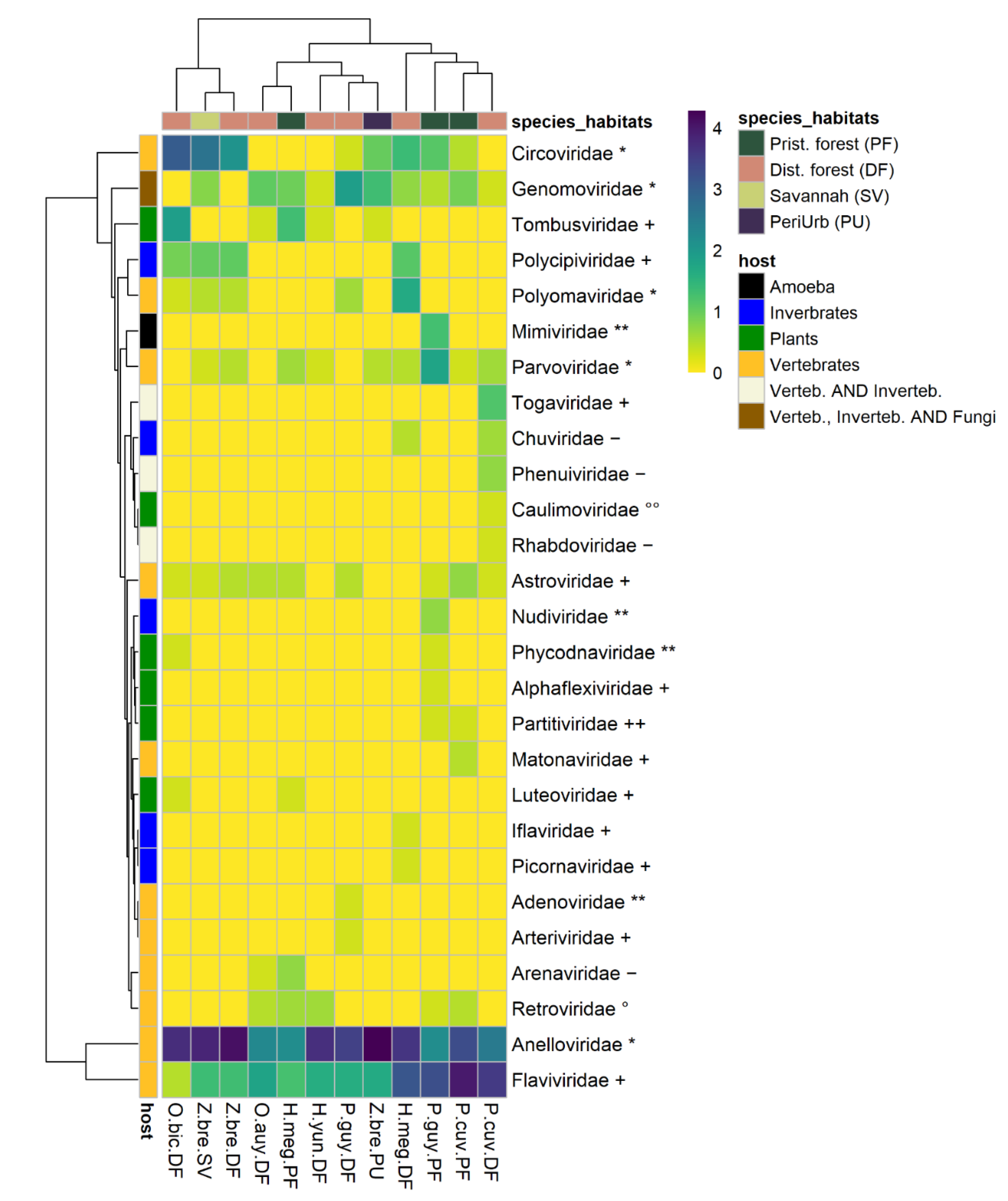
3.2. Viral Diversity Detected through Species and Environments
3.2.1. Plant Viruses
3.2.2. Invertebrate Viruses
3.2.3. Vertebrate Viruses
3.2.4. Potential Vector-Borne Viruses
3.3. Statistical and Ecological Analyses of the Viromes According to Hosts and Environments
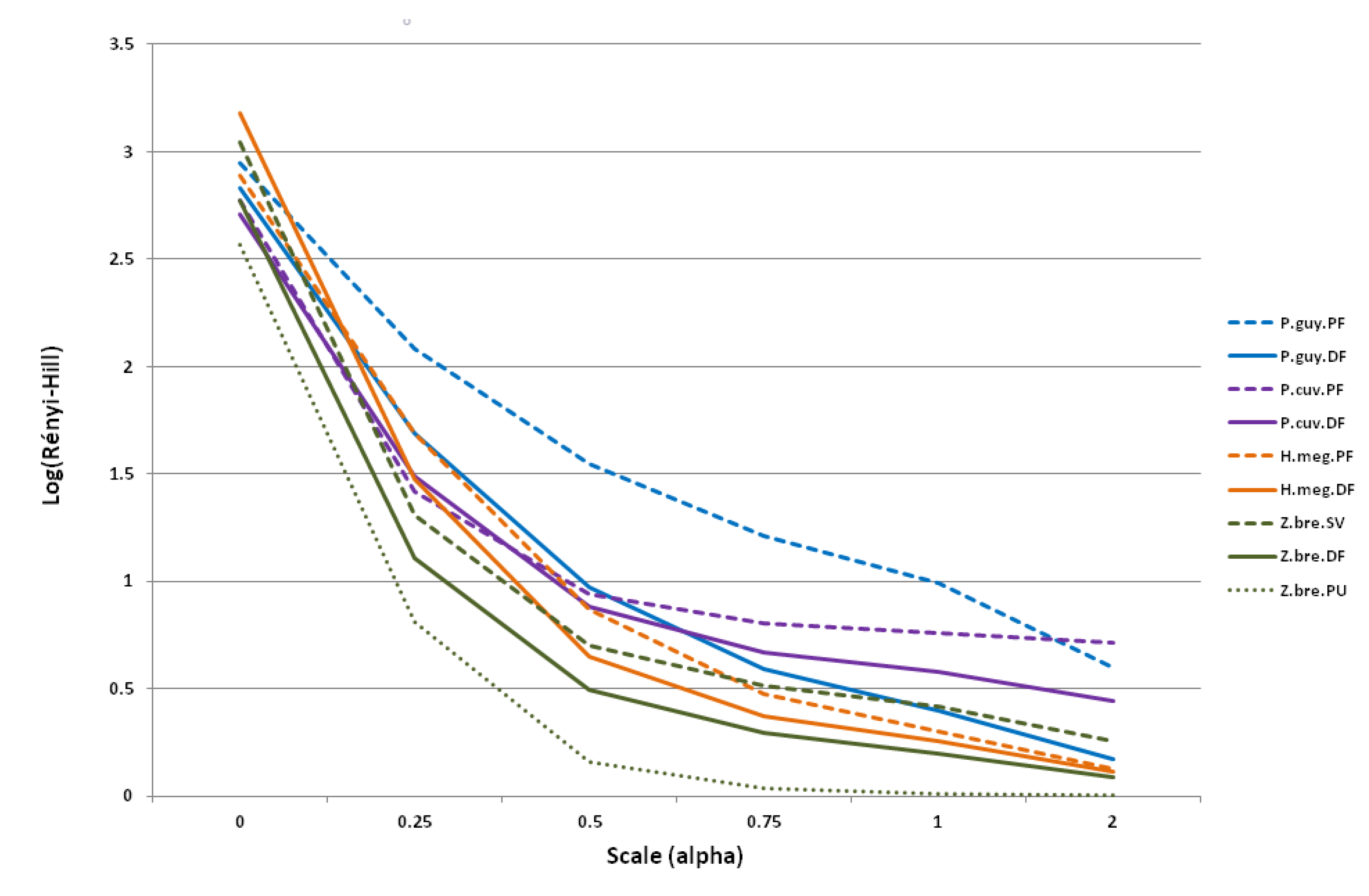
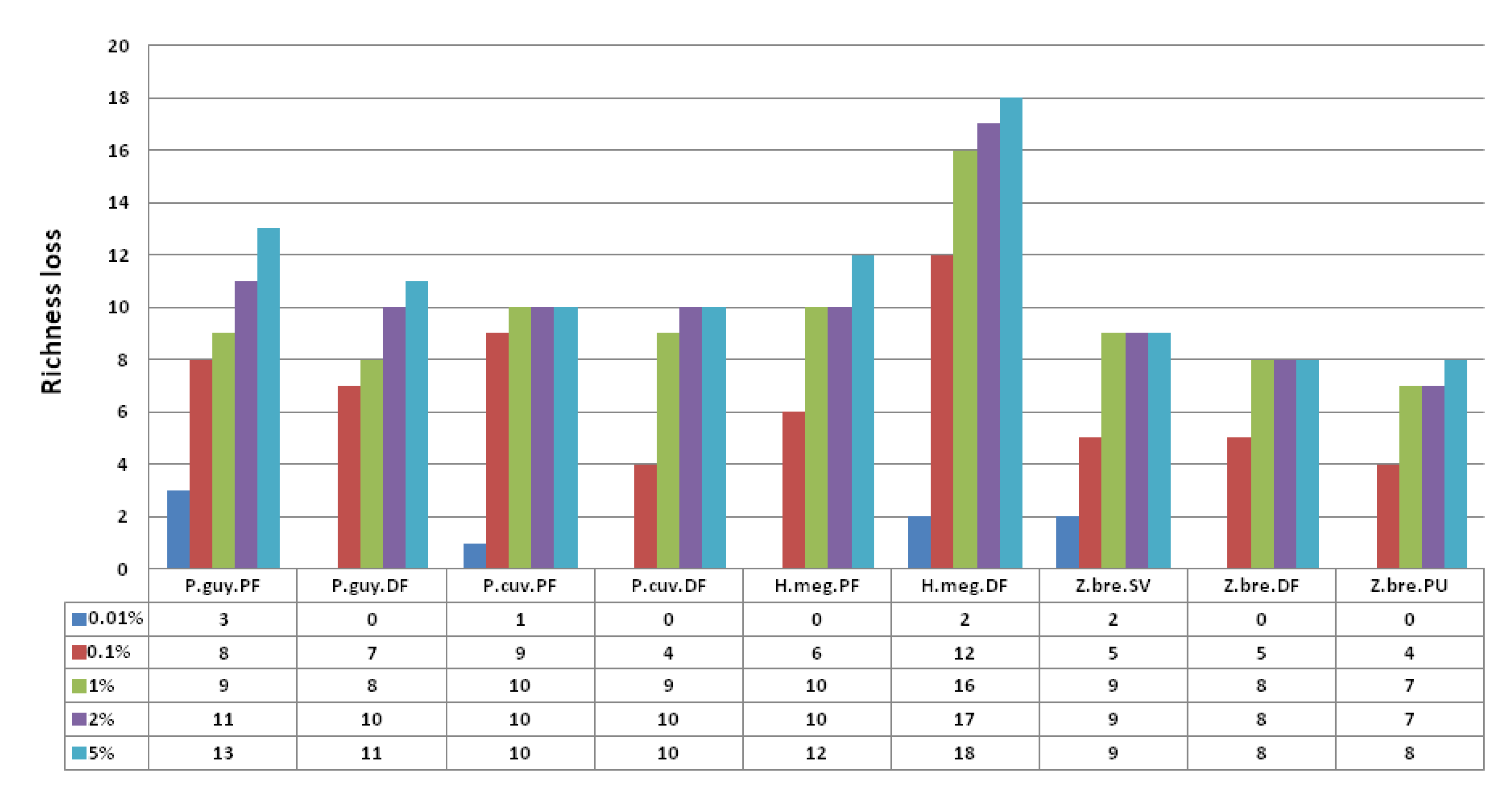
3.3.1. Sampling Effort on Viral Diversity
3.3.2. Diversity across Species–Habitats
3.4. Phylogenetic Relationships of Selected Viruses
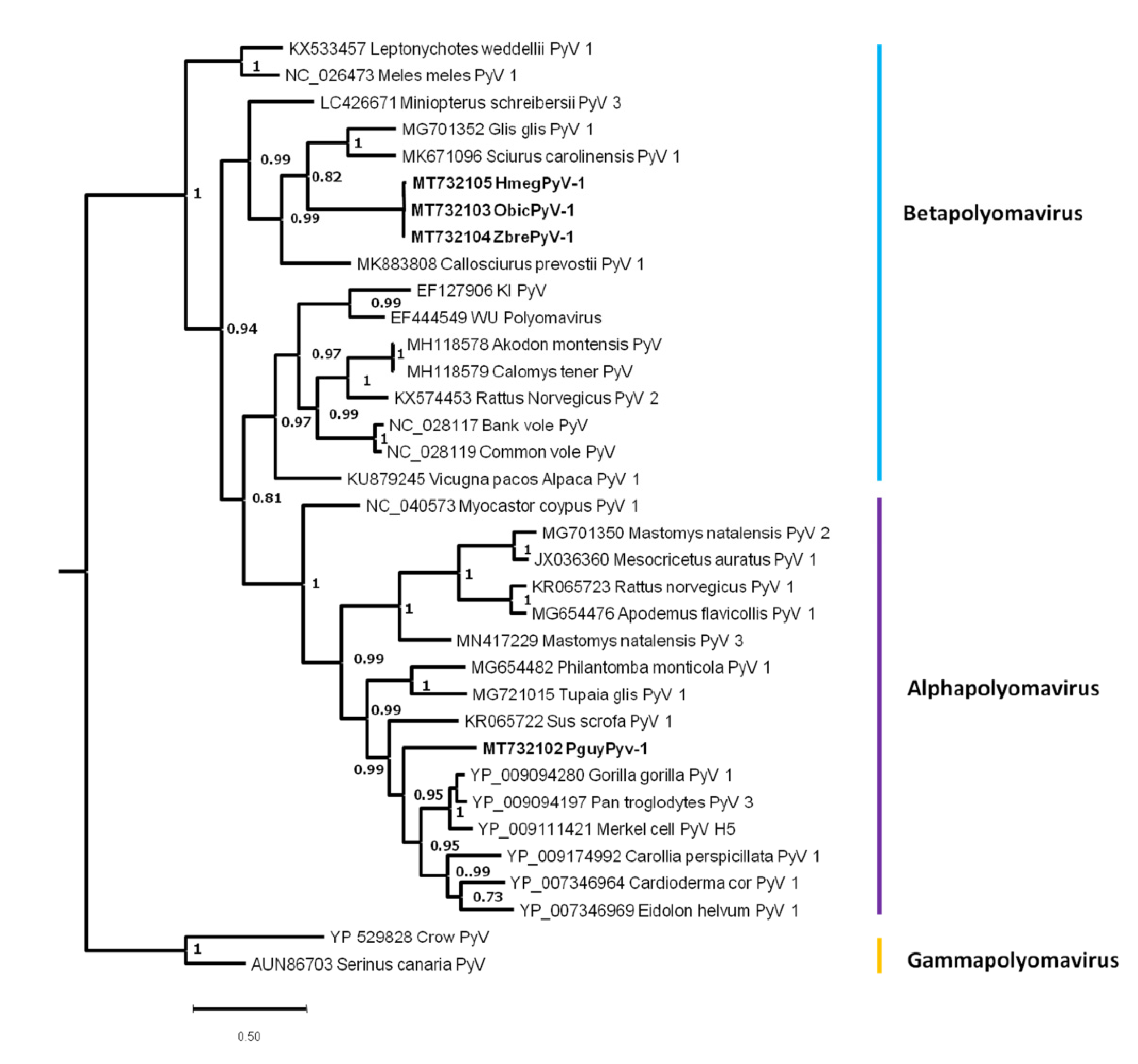
3.4.1. Rodent Polyomaviruses
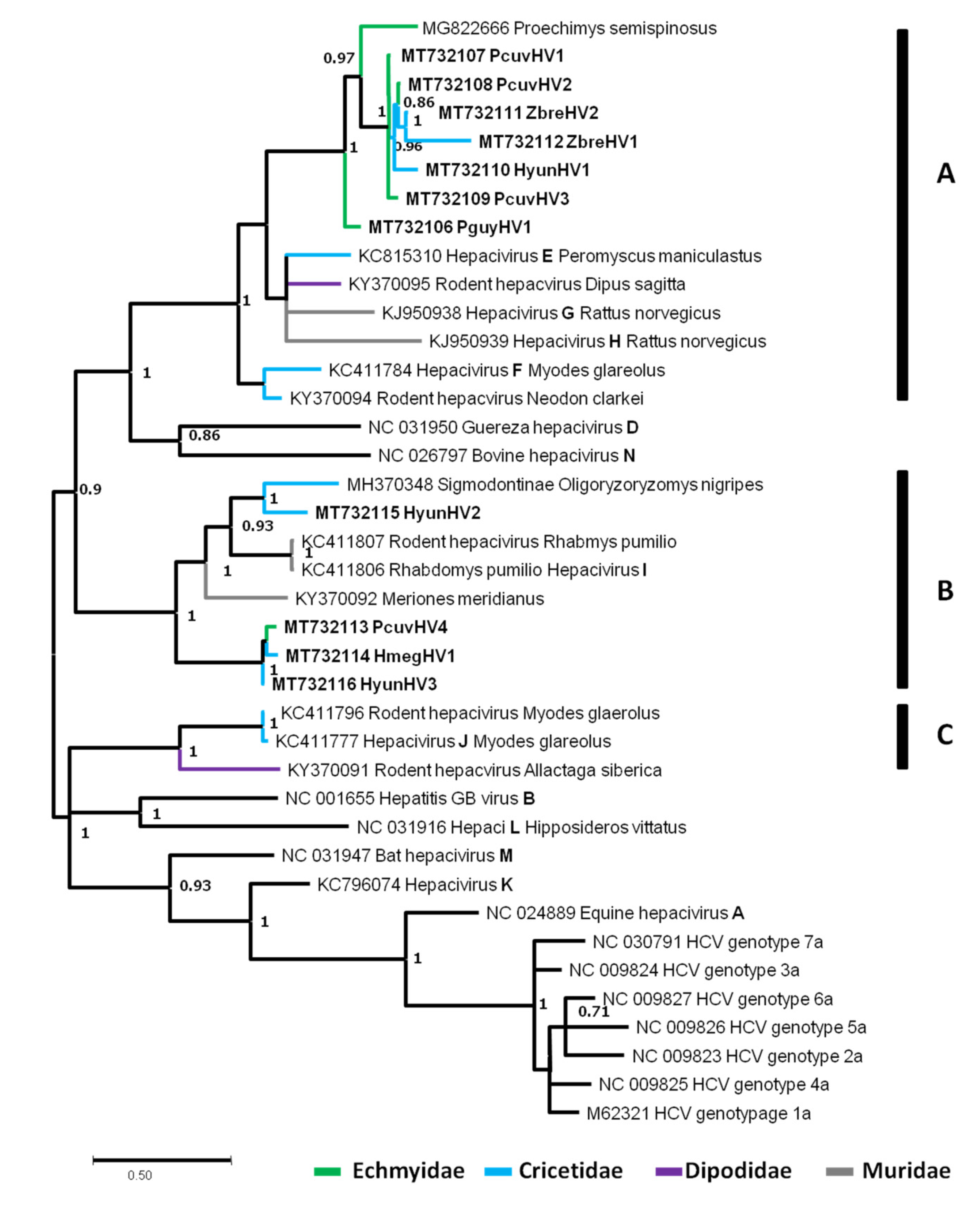
3.4.2. Rodent Hepaciviruses
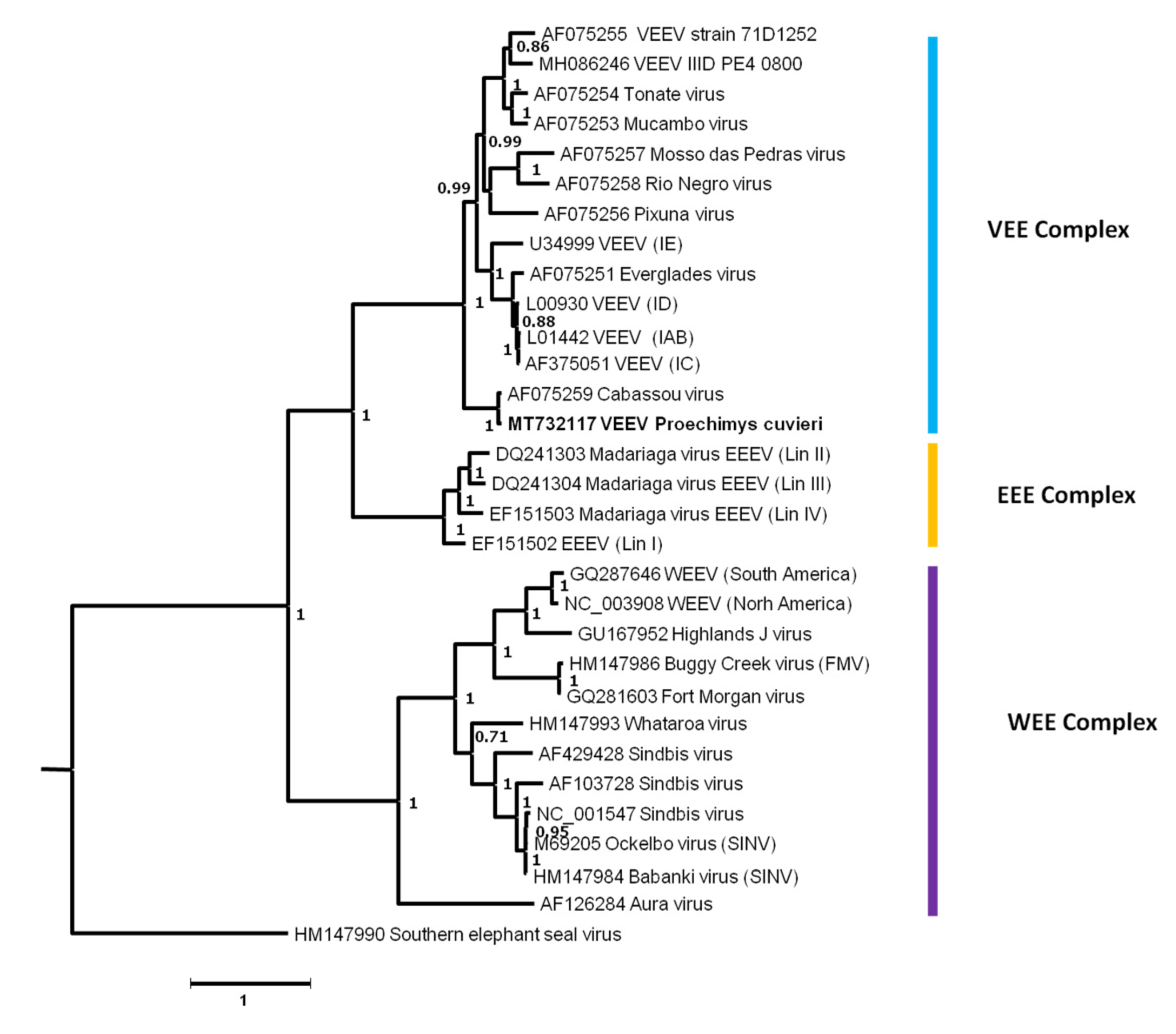
3.4.3. Alphaviruses (Togaviridae)
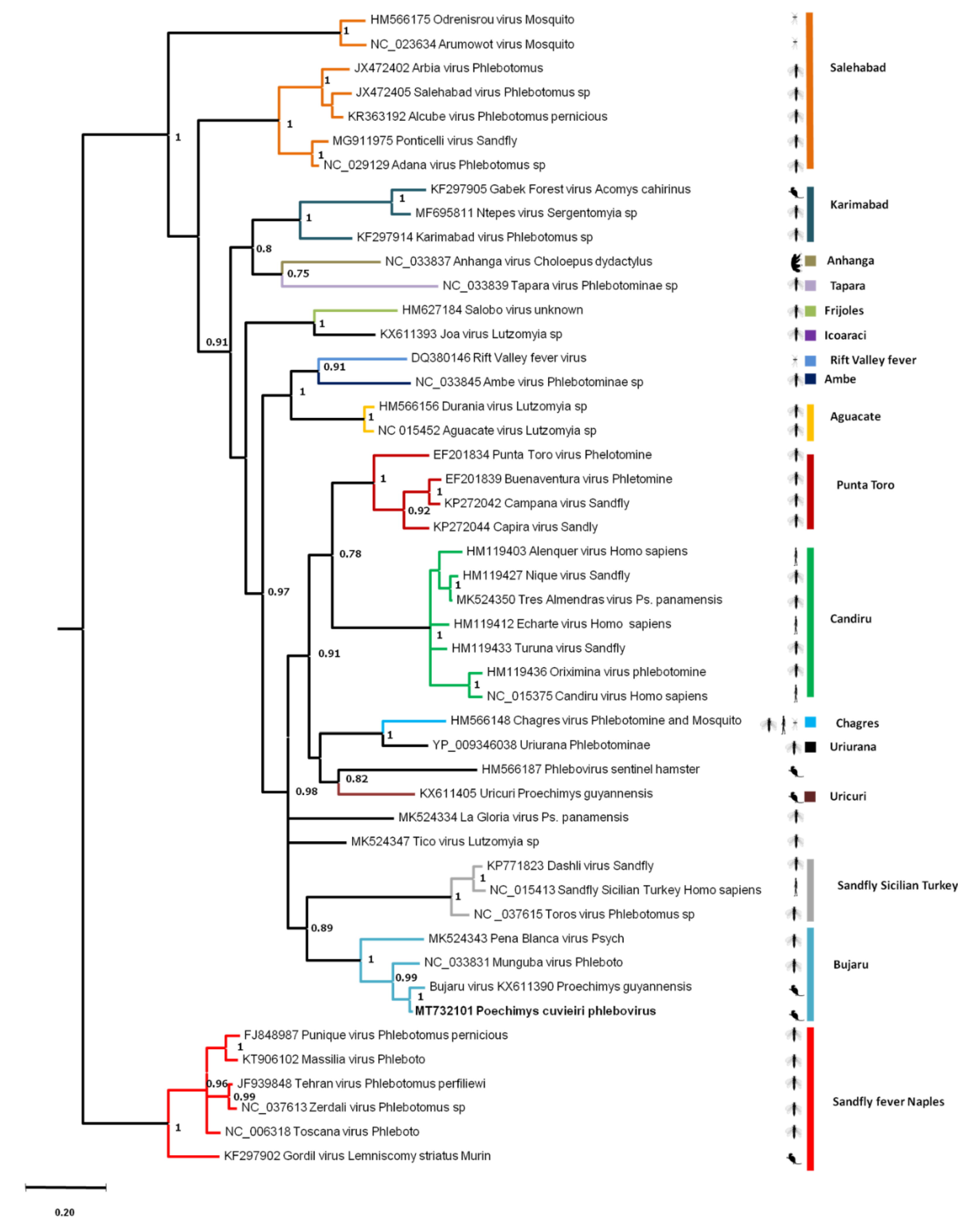
3.4.4. Rodent Phleboviruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral diversity of house mice in New York City. MBio 2018, 9, e02101-19. [Google Scholar] [CrossRef] [Green Version]
- Hartlage, A.S.; Cullen, J.M.; Kapoor, A. The strange, expanding World of animal Hepaciviruses. Annu. Rev. Virol. 2016, 3, 53–75. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The ancient evolutionary history of Polyomaviruses. PLoS Pathog. 2016, 12, e1000863. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergner, L.M.; Orton, R.J.; Benavides, J.A.; Becker, D.J.; Tello, C.; Biek, R.; Streicker, D.G. Demographic and environmental drivers of metagenomic viral diversity in vampire bats. Mol. Ecol. 2020, 29, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as winners in the game of life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Kreuder Johnson, C.; Hitchens, P.L.; Smiley Evans, T.; Goldstein, T.; Thomas, K.; Clements, A.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; Karesh, W.B.; et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennehy, J.J. Evolutionary ecology of virus emergence. Ann. N. Y. Acad. Sci. 2017, 1389, 124–146. [Google Scholar] [CrossRef]
- McMahon, B.J.; Morand, S.; Gray, J.S. Ecosystem change and zoonoses in the Anthropocene. Zoonoses Public Health 2018, 65, 755–765. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Siu, A.; Wong, Y.C.R. Economic Impact of SARS: The case of Hong Kong. Asian Econ. Pap. 2004, 3, 62–83. [Google Scholar] [CrossRef] [Green Version]
- Huber, C.; Finelli, L.; Stevens, W. The economic and social burden of the 2014 Ebola outbreak in West Africa. J. Infect. Dis. 2018, 218, S698–S704. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control COVID-19 Situation Dashboard. Available online: https://qap.ecdc.europa.eu/public/extensions/COVID-19/COVID-19.html#global-overview-tabes (accessed on 20 July 2021).
- Bogich, T.L.; Chunara, R.; Scales, D.; Chan, E.; Pinheiro, L.C.; Chmura, A.A.; Carroll, D.; Daszak, P.; Brownstein, J.S. Preventing pandemics via international development: A systems approach. PLoS Med. 2012, 9, e1001354. [Google Scholar] [CrossRef]
- PREDICT. Available online: https://ohi.vetmed.ucdavis.edu/programs-projects/predict-project/about (accessed on 28 June 2021).
- Daszak, P.; Carroll, D.; Wolfe, N.; Mazet, J. The global virome project. Int. J. Infect. Dis. 2016, 53, 36. [Google Scholar] [CrossRef] [Green Version]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science (80) 2018, 359, 872–874. [Google Scholar] [CrossRef]
- Kruse, H.; Kirkemo, A.M.; Handeland, K. Wildlife as source of zoonotic infections. Emerg. Infect. Dis. 2004, 10, 2067–2072. [Google Scholar] [CrossRef]
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef] [Green Version]
- Corbet, G.B.; Hill, J.E.; Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species Of the World: A Taxonomic and Geographic Reference, 2nd ed.; Smithsonian Institution Press: Washington, DC, USA, 1993; ISBN 1-56098-217-9. [Google Scholar]
- Huchon, D.; Douzery, E.J.P. From the Old World to the New World: A molecular chronicle of the phylogeny and biogeography of hystricognath rodents. Mol. Phylogenetics Evol. 2001, 20, 238–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravinatti, M.L.; Barbosa, C.M.; Soares, R.M.; Gregori, F. Synanthropic rodents as virus reservoirs and transmitters. Rev. Soc. Bras. Med. Trop. 2020, 53, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruedas, L.A.; Salazar-Bravo, J.; Tinnin, D.S.; Armién, B.; Cáceres, L.; García, A.; Díaz, M.A.; Gracia, F.; Suzán, G.; Peters, C.J.; et al. Community ecology of small mammal populations in Panamá following an outbreak of Hantavirus pulmonary syndrome. J. Vector Ecol. 2004, 29, 177–191. [Google Scholar] [PubMed]
- Chen, L.; Liu, B.; Wu, Z.; Jin, Q.; Yang, J. DRodVir: A resource for exploring the virome diversity in rodents. J. Genet. Genomics 2017, 44, 259–264. [Google Scholar] [CrossRef]
- Database of Rodent-Associated Viruses. Available online: http://www.mgc.ac.cn/DRodVir/ (accessed on 20 July 2021).
- Meerburg, B.G.; Singleton, G.R.; Kijlstra, A. Rodent-borne diseases and their risks for public health Rodent. Crit. Rev. Microbiol. 2009, 35, 221–270. [Google Scholar] [CrossRef] [PubMed]
- De Thoisy, B.; Matheus, S.; Catzeflis, F.; Clément, L.; Barrioz, S.; Guidez, A.; Donato, D.; Cornu, J.F.; Brunaux, O.; Guitet, S.; et al. Maripa Hantavirus in French Guiana: Phylogenetic position and predicted spatial distribution of rodent hosts. Am. J. Trop. Med. Hyg. 2014, 90, 988–992. [Google Scholar] [CrossRef]
- Lavergne, A.; Matheus, S.; Catzeflis, F.; Donato, D.; Lacoste, V.; de Thoisy, B. Rodent-borne viruses in French Guiana. Virologie 2017, 21, E12–E27. [Google Scholar] [CrossRef]
- Liu, J.; Liu, D.Y.; Chen, W.; Li, J.L.; Luo, F.; Li, Q.; Ling, J.X.; Liu, Y.Y.; Xiong, H.R.; Ding, X.H.; et al. Genetic analysis of hantaviruses and their rodent hosts in central-south China. Virus Res. 2012, 163, 439–447. [Google Scholar] [CrossRef]
- Monath, T.P.; Newhouse, V.F.; Kemp, G.E.; Setzer, H.W.; Cacciapuoti, A. Lassa virus isolation from Mastomys natalensis rodents during an epidemic in Sierra Leone. Science 1974, 185, 263–265. [Google Scholar] [CrossRef]
- Guégan, J.-F.F.; Ayouba, A.; Cappelle, J.; de Thoisy, B. Forests and emerging infectious diseases: Unleashing the beast within. Environ. Res. Lett. 2020, 15, 083007. [Google Scholar] [CrossRef]
- Gentry, A.H. Tree species richness of upper Amazonian forests. Proc. Natl. Acad. Sci. USA 1988, 85, 156–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwanger, J.H.; Kulmann-Leal, B.; Kaminski, V.L.; Valverde-Villegas, J.M.; DA VEIGA, A.B.G.; Spilki, F.R.; Fearnside, P.M.; Caesar, L.; Giatti, L.L.; Wallau, G.L.; et al. Beyond diversity loss and climate change: Impacts of Amazon deforestation on infectious diseases and public health. Anais da Academia Brasileira de Ciências 2020, 92, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Han, B.A.; Kramer, A.M.; Drake, J.M. Global patterns of zoonotic disease in mammals. Trends Parasitol. 2016, 32, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Salmier, A.; Tirera, S.; de Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PLoS ONE 2017, 12, e0186943. [Google Scholar] [CrossRef] [Green Version]
- Bolatti, E.M.; Zorec, T.M.; Montani, E.; Hošnjak, L.; Chouhy, D.; Casal, P.E.; Barquez, M. American Free-Tailed Bats (Tadarida brasiliensis) and identification of two novel mammalian viruses. Viruses 2020, 12, 422. [Google Scholar] [CrossRef] [Green Version]
- Matheus, S.; Lavergne, A.; de Thoisy, B.; Dussart, P.; Lacoste, V. Complete genome sequence of a novel Hantavirus variant of Rio Mamore Virus, Maripa Virus, from French Guiana. J. Virol. 2012, 86, 5399. [Google Scholar] [CrossRef] [Green Version]
- Matheus, S.; Djossou, F.; Moua, D.; Bourbigot, A.M.; Hommel, D.; Lacoste, V.; Dussart, P.; Lavergne, A. Hantavirus pulmonary syndrome, French Guiana. Emerg. Infect. Dis. 2010, 16, 739–741. [Google Scholar] [CrossRef]
- Matheus, S.; Kallel, H.; Mayence, C.; Bremand, L.; Houcke, S.; Rousset, D.; Lacoste, V.; de Thoisy, B.; Hommel, D.; Lavergne, A. Hantavirus pulmonary syndrome caused by Maripa virus in French Guiana, 2008–2016. Emerg. Infect. Dis. 2017, 23, 1722–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavergne, A.; de Thoisy, B.; Donato, D.; Guidez, A.; Matheus, S.; Catzeflis, F.; Lacoste, V. Patawa Virus, a new Arenavirus hosted by forest rodents in French Guiana. Ecohealth 2015, 12, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, A.; de Thoisy, B.; Tirera, S.; Donato, D.; Bouchier, C.; Catzeflis, F.; Lacoste, V. Identification of lymphocytic choriomeningitis mammarenavirus in house mouse (Mus musculus, Rodentia) in French Guiana. Infect. Genet. Evol. 2016, 37, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Sikes, R.S. 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mammal. 2016, 97, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Direction régionale de l’ONF Guyane. Occupation du sol en 2015 sur la Bande Littorale de la Guyane et son Évolution Entre 2005 et 2015; Office National des Forêts: Biscarrosse, France, 2017. [Google Scholar]
- European Environment Agency. European Environment Agency, Copenhagen Corine Land Cover; EEA: København, Denmark, 1994.
- Ivanova, N.V.; Dewaard, J.R.; Hebert, P.D.N. An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol. Ecol. Notes 2006, 6, 998–1002. [Google Scholar] [CrossRef]
- Borisenko, A.V.; Lim, B.K.; Ivanova, N.V.; Hanner, R.H.; Hebert, P.D.N. DNA barcoding in surveys of small mammal communities: A field study in Suriname. Mol. Ecol. Resour. 2008, 8, 471–479. [Google Scholar] [CrossRef]
- Allander, T.; Emerson, S.U.; Engle, R.E.; Purcell, R.H.; Bukh, J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. USA 2001, 98, 11609–11614. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-C.; Chain, P.S.G. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 366. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2014, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Index of/pub/taxonomy/new_taxdump. Available online: ftp.ncbi.nlm.nih.gov/pub/taxonomy/new_taxdump (accessed on 13 November 2019).
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ (accessed on 9 August 2020).
- ViralZone. Available online: https://viralzone.expasy.org/5576 (accessed on 9 August 2020).
- Research, Laboratory of Chemical Life Science, Kyoto University Bioinfomatics Center, I. for C. Virus-Host DB. Available online: https://www.genome.jp/virushostdb/ (accessed on 9 August 2020).
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.M.; Stevens, M.H.H.; Wagner, H. The vegan package. Community Ecol. Packag. 2008, 3, 190. [Google Scholar]
- Hill, M.O. Diversity and evenness: An unifying notation and its consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Rényi, A. On measures of entropy and information. In Proceedings of the Fourth Berkeley Symposium on Mathematical Statistics and Probability; Contributions to the Theory of Statistics; University of California Press: Berkeley, CA, USA, 1961; Volume 1, pp. 547–561. [Google Scholar]
- Margalef, R. Información y diversidad específica en las comunidades de organismos. Investigación Pesquera 1956, 3, 99–106. [Google Scholar]
- Magurran, A.E.; Henderson, P.A. Explaining the excess of rare species in natural species abundance distributions. Nature 2003, 422, 714–716. [Google Scholar] [CrossRef]
- Tothmeresz, B. Comparison of different methods for diversity ordering. J. Veg. Sci. 1995, 6, 283–290. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV virus taxonomy profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef]
- Smith, D.B.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J. Gen. Virol. 2016, 97, 2894–2907. [Google Scholar] [CrossRef] [PubMed]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [Green Version]
- Marklewitz, M.; Dutari, L.C.; Paraskevopoulou, S.; Page, R.A.; Loaiza, J.R.; Junglen, S. Diverse novel phleboviruses in sandflies from the Panama Canal area, Central Panama. J. Gen. Virol. 2019, 100, 938–949. [Google Scholar] [CrossRef]
- Nunes-Neto, J.P.; De Souza, W.M.; Acrani, G.O.; Romeiro, M.F.; Fumagalli, M.; Vieira, L.C.; De Almeida Medeiros, D.B.; Lima, J.A.; De Lima, C.P.S.; Cardoso, J.F.; et al. Characterization of the bujaru, frijoles and tapara antigenic complexes into the sandfly fever group and two unclassified phleboviruses from Brazil. J. Gen. Virol. 2017, 98, 585–594. [Google Scholar] [CrossRef] [PubMed]
- PhyloPic. Available online: http://phylopic.org (accessed on 27 November 2020).
- Wu, Z.; Han, Y.; Liu, B.; Li, H.; Zhu, G.; Latinne, A.; Dong, J.; Sun, L.; Du, J.; Zhou, S.; et al. Decoding the RNA viromes of rodent lungs provides new visions into the origin and evolution pattern of rodent-borne diseases in Mainland Southeast Asia. Microbiome 2020, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York city. MBio 2014, 5, e01933-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Rabiela, F.; Suzán, G.; Wiratsudakul, A.; Rico-Chávez, O. Viral metacommunities associated to bats and rodents at different spatial scales. Community Ecol. 2018, 19, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Ashley, W.; Gil-Fernandez, M.; Newsome, T.M.; Di Giallonardo, F.; Ortiz-Baez, A.S.; Mahar, J.E.; Towerton, A.L.; Gillings, M.; Holmes, E.C.; et al. Red fox viromes in urban and rural landscapes. Virus Evol. 2020, 6, veaa065. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Giallonardo, F.D.; Wille, M.; Ortiz-Baez, A.S.; Costa, V.A.; Ghaly, T.; Mifsud, J.C.O.; Turnbull, O.M.H.; Bellwood, D.R.; Williamson, J.E.; et al. Host evolutionary history and ecology shape virome composition in fishes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sachsenröder, J.; Braun, A.; Machnowska, P.; Ng, T.F.F.; Deng, X.; Guenther, S.; Bernstein, S.; Ulrich, R.G.; Delwart, E.; Johne, R. Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. J. Gen. Virol. 2014, 95, 2734–2747. [Google Scholar] [CrossRef] [Green Version]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.B.M.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [Green Version]
- Guillotin, M. Rythmes d’activité et régimes alimentaires de Proechimys cuvieri et d’Oryzomys capito velutinus (Rodentia) en forêt guyanaise. Rev. Ecol. (Terre Vie) 1982, 36, 337–371. [Google Scholar]
- Tier, A.; de Carvalho, W.D.; Rostain, S.; Catzeflis, F.; Claessens, O.; Dewynter, M.; McKey, D.; Mustin, K.; Palisse, M.; de Thoisy, B. The amazonian savannas of French Guiana: Cultural and social importance, biodiversity, and conservation challenges. Trop. Conserv. Sci. 2020, 13, 1940082919900471. [Google Scholar] [CrossRef]
- Solbrig, O.T. The diversity of the savanna ecosystem. Ecol. Stud. 1996, 121, 1–27. [Google Scholar] [CrossRef]
- Gubler, D.J. The global emergence/resurgence of arboviral diseases as public health problems. Arch. Med. Res. 2002, 33, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Machalaba, C.; Karesh, W.B. Emerging infectious disease risk: Shared drivers with environmental change. OIE Rev. Sci. Technol. 2017, 36, 435–444. [Google Scholar] [CrossRef]
- Tilman, D.; May, R.M.; Lehman, C.L.; Nowack, M.A. Habitat destruction and the extinction debt revisited. Nature 1994, 371, 65–66. [Google Scholar] [CrossRef]
- Ostfeld, R.S.; Keesing, F. Biodiversity and disease risk: The case of Lyme disease. Conserv. Biol. 2000, 14, 722–728. [Google Scholar] [CrossRef]
- Schmid, J.; Rasche, A.; Eibner, G.; Jeworowski, L.; Page, R.A.; Corman, V.M.; Drosten, C.; Sommer, S. Ecological drivers of Hepacivirus infection in a neotropical rodent inhabiting landscapes with various degrees of human environmental change. Oecologia 2018, 188, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Suzán, G.; Marcé, E.; Giermakowski, J.T.; Mills, J.N.; Ceballos, G.; Ostfeld, R.S.; Armién, B.; Pascale, J.M.; Yates, T.L. Experimental evidence for reduced rodent diversity causing increased hantavirus prevalence. PLoS ONE 2009, 4, e5461. [Google Scholar] [CrossRef] [PubMed]
- Maia, F.G.M.; de Souza, W.M.; Sabino-Santos, G.; Fumagalli, M.J.; Modha, S.; Murcia, P.R.; Figueiredo, L.T.M. A novel polyomavirus in sigmodontine rodents from São Paulo State, Brazil. Arch. Virol. 2018, 163, 2913–2915. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.S.; Cibulski, S.P.; Alves, C.D.B.T.; Weber, M.N.; Budaszewski, R.F.; Silveira, S.; Mósena, A.C.S.; Mayer, F.Q.; Goltz, L.V.; Campos, R.; et al. New polyomavirus species identified in nutria, Myocastor coypus polyomavirus 1. Arch. Virol. 2018, 163, 3203–3206. [Google Scholar] [CrossRef]
- Moens, U.; Krumbholz, A.; Ehlers, B.; Zell, R.; Johne, R.; Calvignac-Spencer, S.; Lauber, C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. [Google Scholar] [CrossRef]
- Calvignac-Spencer, S.; Feltkamp, M.C.W.; Daugherty, M.D.; Moens, U.; Ramqvist, T.; Johne, R.; Ehlers, B. A taxonomy update for the family Polyomaviridae. Arch. Virol. 2016, 161, 1739–1750. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, B.; Anoh, A.E.; Salem, N.B.; Broll, S.; Couacy-Hymann, E.; Fischer, D.; Gedvilaite, A.; Ingenhütt, N.; Liebmann, S.; Martin, M.; et al. Novel polyomaviruses in mammals from multiple orders and reassessment of polyomavirus evolution and taxonomy. Viruses 2019, 11, 930. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Gonzalez, G.; Sheng, J.; Wu, J.; Zhang, F.; Xu, L.; Zhang, P.; Zhu, A.; Qu, Y.; Tu, C.; et al. Extensive genetic diversity of Polyomaviruses in sympatric bat communities: Host switching versus coevolution. J. Virol. 2020, 94, e02101-19. [Google Scholar] [CrossRef]
- Carr, M.; Gonzalez, G.; Sasaki, M.; Dool, S.E.; Ito, K.; Ishii, A.; Hang’ombe, B.M.; Mweene, A.S.; Teeling, E.C.; Hall, W.W.; et al. Identification of the same polyomavirus species in different African horseshoe bat species is indicative of short-range host-switching events. J. Gen. Virol. 2017, 98, 2771–2785. [Google Scholar] [CrossRef] [PubMed]
- Rasche, A.; Sander, A.L.; Corman, V.M.; Drexler, J.F. Evolutionary biology of human hepatitis viruses. J. Hepatol. 2019, 70, 501–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Liu, M.M.; Shen, S.; Zhang, Y.J.; Xu, Y.L.; Deng, H.Y.; Deng, F.; Duan, Z.J. Detection and characterization of a novel hepacivirus in long-tailed ground squirrels (Spermophilus undulatus) in China. Arch. Virol. 2019, 164, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.H.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Sanchez Leon, M.; Jain, K.; et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. MBio 2013, 4, e00216-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for Novel Hepaciviruses in Rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef] [Green Version]
- de Souza, W.; Fumagalli, M.; Sabino-Santos, G.; Motta Maia, F.; Modha, S.; Teixeira Nunes, M.; Murcia, P.; Moraes Figueiredo, L. A novel Hepacivirus in wild rodents from South America. Viruses 2019, 11, 297. [Google Scholar] [CrossRef] [Green Version]
- Scheel, T.K.H.; Simmonds, P.; Kapoor, A. Surveying the global virome: Identification and characterization of HCV-related animal hepaciviruses. Antivir. Res. 2015, 115, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaender, S.; Brown, R.J.P.; Pietschmann, T.; Steinmann, E. Natural reservoirs for homologs of hepatitis C virus. Emerg. Microbes Infect. 2014, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Gray, R.R. The virus whose family expanded. Nature 2013, 498, 310–311. [Google Scholar] [CrossRef]
- Weaver, S.C.; Winegar, R.; Manger, I.D.; Forrester, N.L. Alphaviruses: Population genetics and determinants of emergence. Antivir. Res. 2012, 94, 242–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, P.V.; Greene, I.P.; Coffey, L.L.; Medina, G.; Moncayo, A.C.; Anishchenko, M.; Ludwig, G.V.; Turell, M.J.; O’Guinn, M.L.; Lee, J.; et al. Endemic Venezuelan Equine Encephalitis in northern Peru. Emerg. Infect. Dis. 2004, 10, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R.; Weaver, S.C.; De Siger, J.; Medina, G.; Salas, R.A. Emergence of a new epidemic/epizootic Venezuelan Equine Encephalitis virus in South America. Proc. Natl. Acad. Sci. USA 1995, 92, 5278–5281. [Google Scholar] [CrossRef] [Green Version]
- Rivas, F.; Diaz, L.A.; Cardenas, V.M.; Daza, E.; Bruzon, L.; Alcala, A.; De La Hoz, O.; Caceres, F.M.; Aristizabal, G.; Martinez, J.W.; et al. Epidemic Venezuelan Equine Encephalitis in La Guajira, Colombia, 1995. J. Infect. Dis. 1997, 175, 828–832. [Google Scholar] [CrossRef]
- Taylor, K.G.; Paessler, S. Pathogenesis of Venezuelan Equine Encephalitis. Vet. Microbiol. 2013, 167, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan Equine Encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; O’Brien, L.; Appleton, R.E.; Carr, S.; Bennett, A. Intranasal immunisation with defective adenovirus serotype 5 expressing the Venezuelan Equine Encephalitis virus E2 glycoprotein protects against airborne challenge with virulent virus. Vaccine 2005, 23, 1615–1623. [Google Scholar] [CrossRef]
- Hommel, D.; Heraud, J.M.; Hulin, A.; Talarmin, A. Association of Tonate virus (subtype IIIB of the Venezuelan Equine Encephalitis complex) with encephalitis in a human. Clin. Infect. Dis. 2000, 30, 188–190. [Google Scholar] [CrossRef] [Green Version]
- Monath, T.P.; Lazuick, J.S.; Cropp, C.B.; Rush, W.A.; Calisher, C.H.; Kinney, R.M.; Trent, D.W.; Kemp, G.E.; Bowen, G.S.; Francy, D.B. Recovery of Tonate virus (“bijou bridge” strain), a member of the Venezuelan Equine Encephalomyelitis virus complex, from Cliff Swallow nest bugs (Oeciacus vicarius) and nestling birds in North America. Am. J. Trop. Med. Hyg. 1980, 29, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Carrara, A.S.; Gonzales, M.; Ferro, C.; Tamayo, M.; Aronson, J.; Paessler, S.; Anishchenko, M.; Boshell, J.; Weaver, S.C. Venezuelan Equine Encephalitis virus infection of spiny rats. Emerg. Infect. Dis. 2005, 11, 663–669. [Google Scholar] [CrossRef]
- Deardorff, E.R.; Forrester, N.L.; Travassos Da Rosa, A.P.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Tesh, R.B.; Weaver, S.C. Experimental infection of potential reservoir hosts with Venezuelan Equine Encephalitis virus, Mexico. Emerg. Infect. Dis. 2009, 15, 519–525. [Google Scholar] [CrossRef]
- Mutricy, R.; Djossou, F.; Matheus, S.; Lorenzi-Martinez, E.; De Laval, F.; Demar, M.; Nacher, M.; Rousset, D.; Epelboin, L. Discriminating Tonate Virus from Dengue virus infection: A matched case–control study in French Guiana, 2003–2016. Am. J. Trop. Med. Hyg. 2020, 102, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, P.V.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.D.; Weaver, S.C. Endemic Venezuelan Equine Encephalitis in the Americas: Hidden under the dengue umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Carvalho, M.S.; De Lara Pinto, A.Z.; Pinheiro, A.; Rodrigues, J.S.V.; Melo, F.L.; Da Silva, L.A.; Ribeiro, B.M.; Dezengrini-Slhessarenko, R. Viola Phlebovirus is a novel Phlebotomus fever serogroup member identified in Lutzomyia (Lutzomyia) longipalpis from Brazilian Pantanal. Parasites Vectors 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, G.; Wiley, M.R.; Travassos da Rosa, A.P.A.; Guzman, H.; Quiroz, E.; Savji, N.; Carrera, J.P.; Bussetti, A.V.; Ladner, J.T.; Ian Lipkin, W.; et al. Characterization of the Punta Toro species complex (genus Phlebovirus, family Bunyaviridae). J. Gen. Virol. 2015, 96, 2079–2085. [Google Scholar] [CrossRef]
- Gundacker, N.D.; Carrera, J.P.; Castillo, M.; Díaz, Y.; Valenzuela, J.; Tamhane, A.; Moreno, B.; Pascale, J.M.; Tesh, R.B.; López-Vergès, S. Clinical manifestations of Punta Toro virus species complex infections, Panama, 2009. Emerg. Infect. Dis. 2017, 23, 872–874. [Google Scholar] [CrossRef]
- Lorenz, C.; de Oliveira Lage, M.; Chiaravalloti-Neto, F. Deforestation hotspots, climate crisis, and the perfect scenario for the next epidemic: The Amazon time bomb. Sci. Total Environ. 2021, 783, 147090. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool Number | Sample ID | Organ | Species | Habitat | Nb. Individuals | Total of Different Individuals |
|---|---|---|---|---|---|---|
| 1 | k_Pguy_PF | Kidney | Proechimys guyannensis | Pristine Forest | 5 | 12 |
| 2 | r_Pguy_PF | Spleen | Proechimys guyannensis | Pristine Forest | 8 | |
| 3 | s_Pguy_PF | Sera | Proechimys guyannensis | Pristine Forest | 12 | |
| 4 | k_Pguy_DF | Kidney | Proechimys guyannensis | Disturbed Forest | 24 | 29 |
| 5 | r_Pguy_DF | Spleen | Proechimys guyannensis | Disturbed Forest | 27 | |
| 6 | s_Pguy_DF | Sera | Proechimys guyannensis | Disturbed Forest | 27 | |
| 7 | k_Pcuv_PF | Kidney | Proechimys cuvieri | Pristine Forest | 9 | 35 |
| 8 | r_Pcuv_PF | Spleen | Proechimys cuvieri | Pristine Forest | 24 | |
| 9 | s_Pcuv_PF | Sera | Proechimys cuvieri | Pristine Forest | 31 | |
| 10 | k_Pcuv_DF | Kidney | Proechimys cuvieri | Disturbed Forest | 18 | 18 |
| 11 | r_Pcuv_DF | Spleen | Proechimys cuvieri | Disturbed Forest | 18 | |
| 12 | s_Pcuv_DF | Sera | Proechimys cuvieri | Disturbed Forest | 15 | |
| 13 | k_Oauy_DF | Kidney | Oecomys auyantepui | Disturbed Forest | 5 | 14 |
| 14 | r_Oauy_DF | Spleen | Oecomys auyantepui | Disturbed Forest | 11 | |
| 15 | s_Oauy_DF | Sera | Oecomys auyantepui | Disturbed Forest | 13 | |
| 16 | k_Obic_DF | Kidney | Oecomys bicolor | Disturbed Forest | 13 | 15 |
| 17 | r_Obic_DF | Spleen | Oecomys bicolor | Disturbed Forest | 14 | |
| 18 | s_Obic_DF | Sera | Oecomys bicolor | Disturbed Forest | 12 | |
| 19 | k_Hyun_DF | Kidney | Hylaeamys yunganus | Disturbed Forest | 11 | 11 |
| 20 | r_Hyun_DF | Spleen | Hylaeamys yunganus | Disturbed Forest | 11 | |
| 21 | s_Hyun_DF | Sera | Hylaeamys yunganus | Disturbed Forest | 10 | |
| 22 | k_Hmeg_PF | Kidney | Hylaeamys megacephalus | Pristine Forest | 2 | 5 |
| 23 | r_Hmeg_PF | Spleen | Hylaeamys megacephalus | Pristine Forest | 4 | |
| 24 | s_Hmeg_PF | Sera | Hylaeamys megacephalus | Pristine Forest | 3 | |
| 25 | k_Hmeg_DF | Kidney | Hylaeamys megacephalus | Disturbed Forest | 4 | 5 |
| 26 | r_Hmeg_DF | Spleen | Hylaeamys megacephalus | Disturbed Forest | 4 | |
| 27 | s_Hmeg_DF | Sera | Hylaeamys megacephalus | Disturbed Forest | 4 | |
| 28 | k_Zbre_SV | Kidney | Zygodontomys brevicauda | Savannah | 6 | 12 |
| 29 | r_Zbre_SV | Spleen | Zygodontomys brevicauda | Savannah | 6 | |
| 30 | s_Zbre_SV | Sera | Zygodontomys brevicauda | Savannah | 8 | |
| 31 | k_Zbre_DF | Kidney | Zygodontomys brevicauda | Disturbed Forest | 7 | 10 |
| 32 | r_Zbre_DF | Spleen | Zygodontomys brevicauda | Disturbed Forest | 7 | |
| 33 | s_Zbre_DF | Sera | Zygodontomys brevicauda | Disturbed Forest | 10 | |
| 34 | k_Zbre_PU | Kidney | Zygodontomys brevicauda | Peri-urban | 20 | 21 |
| 35 | r_Zbre_PU | Spleen | Zygodontomys brevicauda | Peri-urban | 20 | |
| 36 | s_Zbre_PU | Sera | Zygodontomys brevicauda | Peri-urban | 19 | |
| Totals | - | - | - | 442 | 187 |
| Sample ID | No. of Raw Reads | Sequencing Type | % Cleaned Reads | No. of Contigs | % Mapped Reads | No. of Viral Contigs |
|---|---|---|---|---|---|---|
| k_Pguy_PF | 39,505,174 | Hiseq2500 | 98.12 | 323,113 | 97.81 | 101 |
| k_Pguy_DF | 24,142,444 | Hiseq2500 | 98.99 | 143,955 | 97.88 | 161 |
| k_Pcuv_PF | 39,878,026 | Hiseq2500 | 98.15 | 267,539 | 98.33 | 398 |
| k_Pcuv_DF | 32,782,446 | Hiseq2500 | 99.56 | 88,325 | 99.30 | 4783 |
| k_Oauy_DF | 43,211,478 | Hiseq2500 | 98.01 | 225,793 | 98.36 | 86 |
| k_Obic_DF | 3,999,474 | MiSeq | 97.55 | 31,337 | 98.92 | 2208 |
| k_Hmeg_PF | 35,419,066 | Hiseq2500 | 98.50 | 285,067 | 97.96 | 71 |
| k_Hmeg_DF | 6,259,058 | MiSeq | 96.62 | 53,664 | 98.99 | 2998 |
| k_Hyun_DF | 31,133,790 | Hiseq2500 | 98.48 | 159,779 | 98.77 | 142 |
| k_Zbre_PU | 34,912,952 | Hiseq2500 | 98.48 | 181,251 | 98.79 | 150 |
| k_Zbre_DF | 6,754,874 | MiSeq | 98.35 | 56,113 | 98.75 | 1698 |
| k_Zbre_SV | 4,395,890 | MiSeq | 98.16 | 25,215 | 99.25 | 1656 |
| r_Pguy_PF | 40,349,120 | Hiseq2500 | 98.01 | 330,777 | 97.87 | 79 |
| r_Pguy_DF | 29,988,950 | Hiseq2500 | 98.22 | 231,108 | 97.69 | 125 |
| r_Pcuv_PF | 16,840,948 | Hiseq2500 | 98.77 | 124,874 | 97.58 | 384 |
| r_Pcuv_DF | 27,294,898 | Hiseq2500 | 98.28 | 267,976 | 97.05 | 909 |
| r_Oauy_DF | 33,188,412 | Hiseq2500 | 98.57 | 184,913 | 98.19 | 109 |
| r_Obic_DF | 5,964,414 | MiSeq | 98.74 | 67,976 | 98.40 | 3506 |
| r_Hmeg_PF | 35,586,626 | Hiseq2500 | 98.22 | 282,402 | 98.10 | 121 |
| r_Hmeg_DF | 7,633,778 | MiSeq | 97.32 | 53,117 | 99.38 | 1109 |
| r_Hyun_DF | 33,617,438 | Hiseq2500 | 98.54 | 159,882 | 98.40 | 317 |
| r_Zbre_PU | 44,909,868 | Hiseq2500 | 99.02 | 176,663 | 99.18 | 13,343 |
| r_Zbre_DF | 5,029,582 | MiSeq | 97.65 | 59,922 | 98.53 | 2591 |
| r_Zbre_SV | 4,296,504 | MiSeq | 95.59 | 79,251 | 97.40 | 617 |
| s_Pguy_PF | 20,326,680 | Hiseq2500 | 98.86 | 56,461 | 98.83 | 2115 |
| s_Pguy_DF | 25,433,042 | Hiseq2500 | 99.49 | 54,835 | 99.01 | 3473 |
| s_Pcuv_PF | 30,224,896 | Hiseq2500 | 98.99 | 86,555 | 99.30 | 12,162 |
| s_Pcuv_DF | 25,361,476 | Hiseq2500 | 98.15 | 347,957 | 96.80 | 3341 |
| s_Oauy_DF | 18,055,176 | Hiseq2500 | 98.00 | 71,707 | 98.84 | 255 |
| s_Obic_DF | 26,895,348 | Hiseq2500 | 99.23 | 103,205 | 98.55 | 2114 |
| s_Hmeg_PF | 32,497,436 | Hiseq2500 | 99.48 | 75,533 | 99.19 | 248 |
| s_Hmeg_DF | 22,859,508 | Hiseq2500 | 98.62 | 70,539 | 99.04 | 1862 |
| s_Hyun_DF | 24,873,840 | Hiseq2500 | 98.78 | 258,141 | 96.09 | 6109 |
| s_Zbre_PU | 26,110,716 | Hiseq2500 | 99.35 | 87,791 | 98.60 | 10,330 |
| s_Zbre_DF | 37,050,082 | Hiseq2500 | 99.09 | 103,974 | 99.07 | 13,622 |
| s_Zbre_SV | 30,948,566 | Hiseq2500 | 98.94 | 91,402 | 98.89 | 8574 |
| Total | 907,731,976 | - | - | 5,268,112 | - | 101,867 |
| P.guy.PF | P.guy.DF | P.cuv.PF | P.cuv.DF | H.meg.PF | H.meg.DF | Z.bre.SV | Z.bre.DF | Z.bre.PU | |
|---|---|---|---|---|---|---|---|---|---|
| Richness | 17 | 16 | 14 | 12 | 13 | 21 | 12 | 11 | 10 |
| Renyi (α = 0) | 2.94 | 2.83 | 2.77 | 2.71 | 2.89 | 3.18 | 3.04 | 2.77 | 2.56 |
| Renyi (α = 0.25) | 2.08 | 1.69 | 1.42 | 1.49 | 1.69 | 1.47 | 1.31 | 1.11 | 0.81 |
| Renyi (α = 0.5) | 1.55 | 0.97 | 0.94 | 0.89 | 0.87 | 0.65 | 0.70 | 0.49 | 0.16 |
| Renyi (α = 0.75) | 1.21 | 0.59 | 0.81 | 0.67 | 0.48 | 0.37 | 0.51 | 0.29 | 0.04 |
| Renyi (α = 1) | 0.99 | 0.40 | 0.76 | 0.58 | 0.30 | 0.25 | 0.42 | 0.20 | 0.01 |
| Renyi (α = 2) | 0.60 | 0.17 | 0.72 | 0.44 | 0.13 | 0.12 | 0.25 | 0.09 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirera, S.; de Thoisy, B.; Donato, D.; Bouchier, C.; Lacoste, V.; Franc, A.; Lavergne, A. The Influence of Habitat on Viral Diversity in Neotropical Rodent Hosts. Viruses 2021, 13, 1690. https://doi.org/10.3390/v13091690
Tirera S, de Thoisy B, Donato D, Bouchier C, Lacoste V, Franc A, Lavergne A. The Influence of Habitat on Viral Diversity in Neotropical Rodent Hosts. Viruses. 2021; 13(9):1690. https://doi.org/10.3390/v13091690
Chicago/Turabian StyleTirera, Sourakhata, Benoit de Thoisy, Damien Donato, Christiane Bouchier, Vincent Lacoste, Alain Franc, and Anne Lavergne. 2021. "The Influence of Habitat on Viral Diversity in Neotropical Rodent Hosts" Viruses 13, no. 9: 1690. https://doi.org/10.3390/v13091690
APA StyleTirera, S., de Thoisy, B., Donato, D., Bouchier, C., Lacoste, V., Franc, A., & Lavergne, A. (2021). The Influence of Habitat on Viral Diversity in Neotropical Rodent Hosts. Viruses, 13(9), 1690. https://doi.org/10.3390/v13091690