
Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Merkel Cell Carcinoma (MCC)
1.2. Merkel Cell Polyomavirus (MCPyV)
1.2.1. MCPyV Discovery
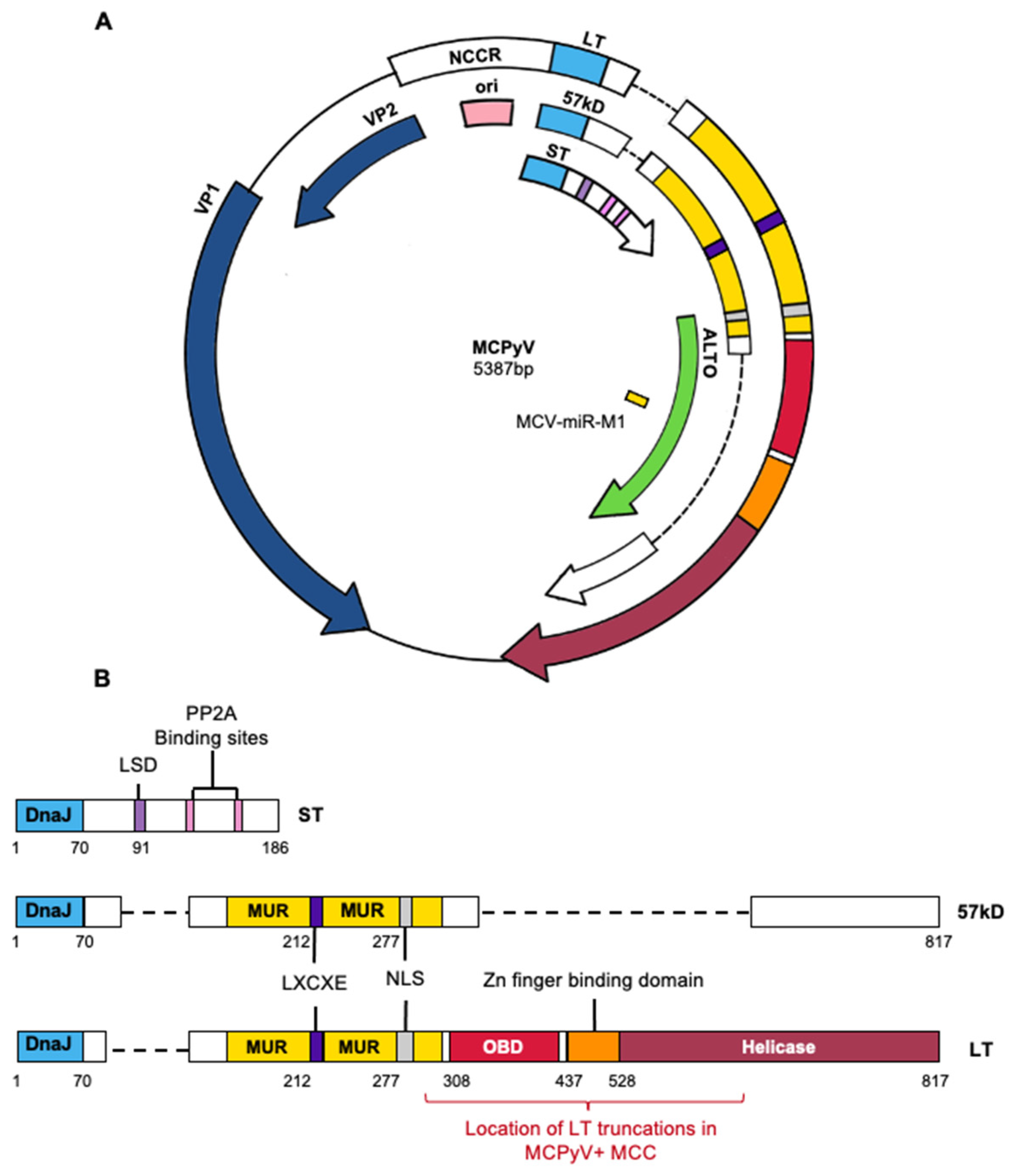
1.2.2. Viral Genome Organization and Viral Gene Products
1.2.3. MCPyV Tropism
2. Role of MCPyV in MCC
2.1. MCC Cell of Origin
2.2. Viral Genome Integration
2.3. MCPyV T Antigen Expression and Oncogenic Properties
2.3.1. MCPyV LT
2.3.2. MCPyV ST
2.4. MCPyV-Associated Cellular Plasticity and/or Reprogramming
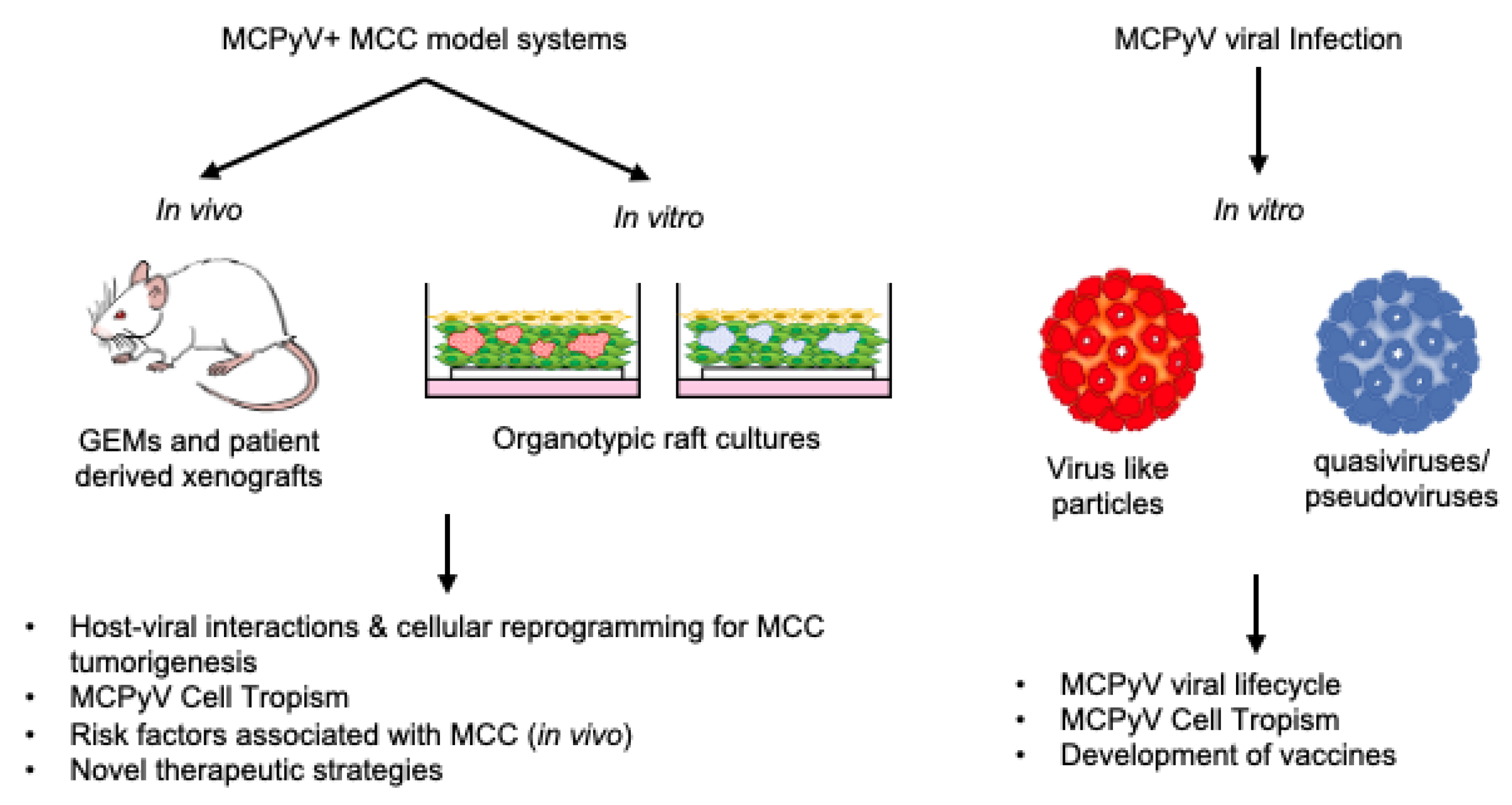
3. Considerations for Designing MCPyV+ MCC Models
4. In Vitro Models of MCPyV Infection & MCPyV+ MCC
4.1. Models of MCPyV Tropism and Infection
4.2. In Vitro Models to Explore MCC Cell of Origin
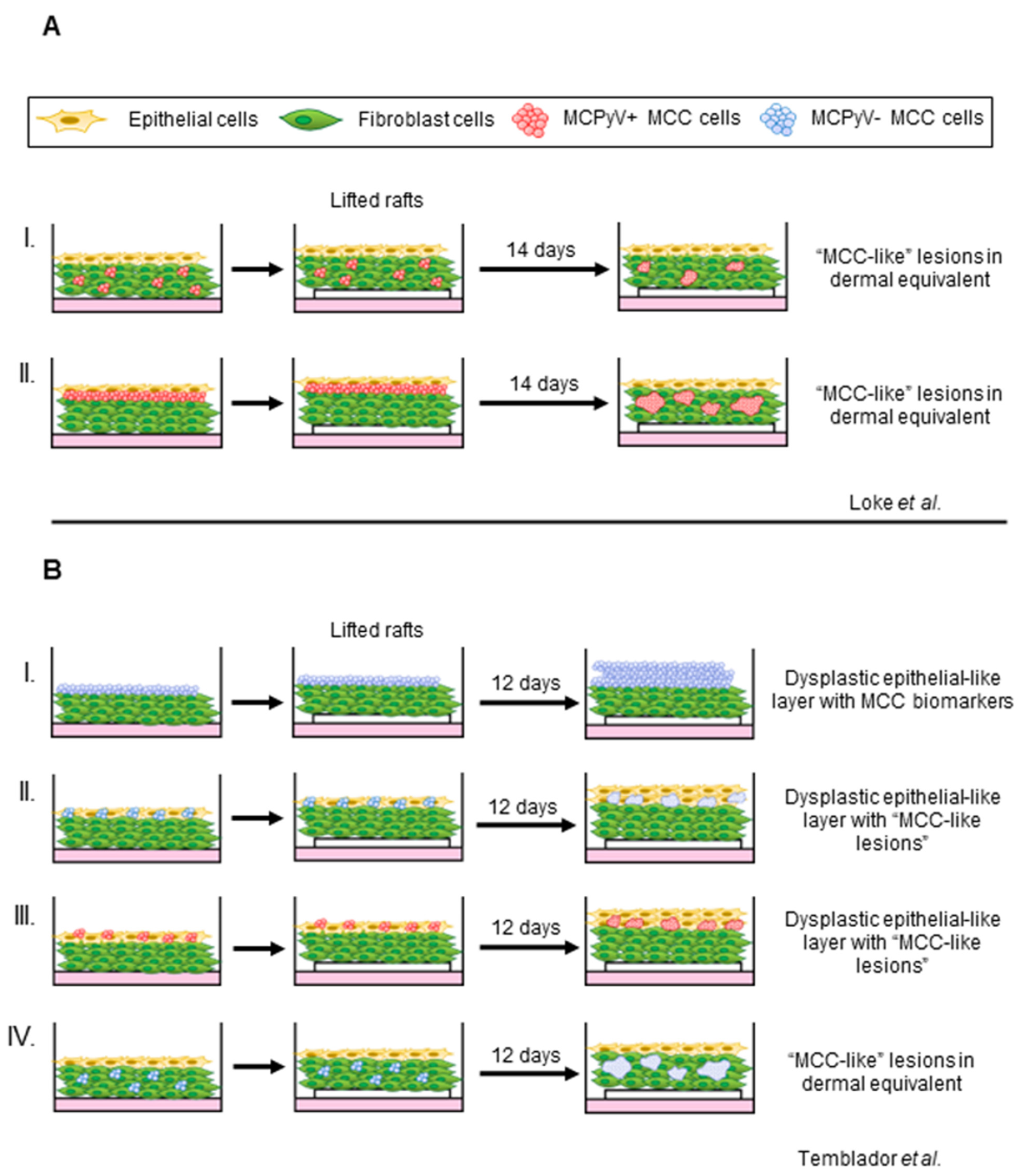
4.3. Organotypic Raft Models of MCPyV+ MCC
5. In Vivo Models of MCPyV+ MCC
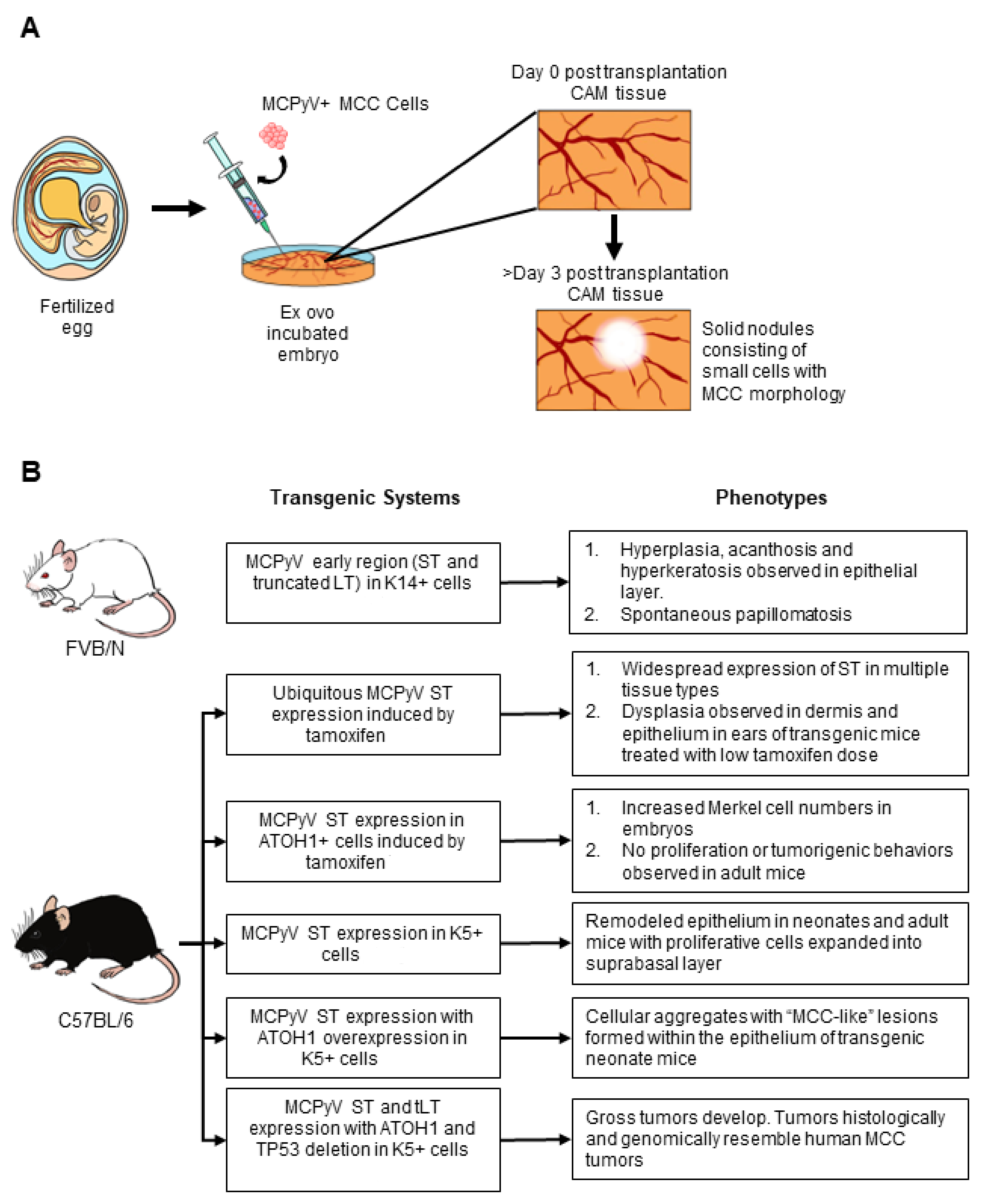
5.1. Chorioallantoic Membrane (CAM) Models of MCPyV+ MCC
5.2. Xenografts and Patient-Derived Xenografts
5.3. Genetically Engineered Mouse Models of MCPyV+ MCC
5.3.1. MCPyV T Antigen GEMs
5.3.2. Combined Host Factor and MCPyV T Antigen GEMs
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles In Cultured Lymphoblasts From Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [Green Version]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Shenk, T. The sequence 5’-AAUAAA-3’forms parts of the recognition site for polyadenylation of late SV40 mRNAs. Cell 1981, 24, 251–260. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad.Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Kelly, T.J. Simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci. USA 1984, 81, 6973–6977. [Google Scholar] [CrossRef] [Green Version]
- Waga, S.; Bauer, G.; Stillman, B. Reconstitution of complete SV40 DNA replication with purified replication factors. J. Biol. Chem. 1994, 269, 10923–10934. [Google Scholar] [CrossRef]
- Lanford, R.E.; Butel, J.S. Construction and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell 1984, 37, 801–813. [Google Scholar] [CrossRef]
- DiMaio, D. Small size, big impact: How studies of small DNA tumour viruses revolutionized biology. Philos. Trans. R. Soc. B 2019, 374, 20180300. [Google Scholar] [CrossRef] [Green Version]
- Howley, P.M.; Livingston, D.M. Small DNA tumor viruses: Large contributors to biomedical sciences. Virology 2009, 384, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Lambert, P.F. Small viruses, big discoveries: The interwoven story of the small DNA tumor viruses. Virology 2009, 384, 255–414. [Google Scholar] [CrossRef] [Green Version]
- Pipas, J.M. DNA tumor viruses and their contributions to molecular biology. J. Virol. 2019, 93, e01524-18. [Google Scholar] [CrossRef] [Green Version]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397. [Google Scholar] [CrossRef] [Green Version]
- Rous, P. A transmissible avian neoplasm.(Sarcoma of the common fowl) by Peyton Rous, MD, Experimental Medicine for Sept. 1, 1910, vol. 12, pp. 696–705. J. Exp. Med. 1979, 150, 729–753. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- DeCaprio, J.A. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortes-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Kouzmina, M.; Koljonen, V.; Leikola, J.; Böhling, T.; Lantto, E. Frequency and locations of systemic metastases in Merkel cell carcinoma by imaging. Acta Radiol. Open 2017, 6, 2058460117700449. [Google Scholar] [CrossRef]
- Lewis, C.W.; Qazi, J.; Hippe, D.S.; Lachance, K.; Thomas, H.; Cook, M.M.; Juhlin, I.; Singh, N.; Thuesmunn, Z.; Takagishi, S.R. Patterns of distant metastases in 215 Merkel cell carcinoma patients: Implications for prognosis and surveillance. Cancer Med. 2020, 9, 1374–1382. [Google Scholar] [CrossRef]
- Toker, C. Trabecular Carcinoma of the Skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Tang, C.K.; Toker, C. Trabecular carcinoma of the skin. An ultrastructural study. Cancer 1978, 42, 2311–2321. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin: A question of title. Am. J. Dermatopathol. 1982, 4, 497–500. [Google Scholar] [CrossRef]
- Rywlin, A. Malignant Merkel-cell tumor is a more accurate description than trabecular carcinoma. Am. J. Dermatopathol. 1982, 4, 513–516. [Google Scholar] [CrossRef]
- Koljonen, V.; Haglund, C.; Tukiainen, E.; Böhling, T. Neuroendocrine differentiation in primary Merkel cell carcinoma-possible prognostic significance. Anticancer Res. 2005, 25, 853–858. [Google Scholar]
- Gallego, R.; García-Caballero, T.; Beiras, A.; Fraga, M.; Forteza, J. Neural cell adhesion molecule immunoreactivity in Merkel cells and Merkel cell tumours. Virchows Arch. 1995, 426, 317–321. [Google Scholar] [CrossRef]
- Lilo, M.T.; Chen, Y.; LeBlanc, R.E. INSM1 is more sensitive and interpretable than conventional immunohistochemical stains used to diagnose Merkel cell carcinoma. Am. J. Surg. Pathol. 2018, 42, 1541–1548. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; McGrath, J.P.; Lim, M.Y.; Cushman, C.; Swanson, S.K.; Tillgren, M.L.; Paulo, J.A.; Gokhale, P.C.; Florens, L. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 2020, 22, 603–615. [Google Scholar] [CrossRef]
- Lucarz, A.; Brand, G. Current considerations about Merkel cells. Eur. J. Cell Biol. 2007, 86, 243–251. [Google Scholar] [CrossRef]
- Moll, I.; Roessler, M.; Brandner, J.M.; Eispert, A.-C.; Houdek, P.; Moll, R. Human Merkel cells–aspects of cell biology, distribution and functions. Eur. J. Cell Biol. 2005, 84, 259–271. [Google Scholar] [CrossRef]
- Sidhu, G.S.; Chandra, P.; Cassai, N.D. Merkel cells, normal and neoplastic: An update. Ultrastruct. Pathol. 2005, 29, 287–294. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermato. 2008, 58, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Gooptu, C.; Woollons, A.; Ross, J.; Price, M.; Wojnarowska, F.; Morris, P.J.; Wall, S.; Bunker, C.B. Merkel cell carcinoma arising after therapeutic immunosuppression. Br. J. Dermatol. 1997, 137, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e452. [Google Scholar] [CrossRef]
- Freeman, M.B.; Holman, D.M.; Qin, J.; Lunsford, N.B. Merkel cell carcinoma incidence, trends, and survival rates among adults aged ≥ 50 years from United States Cancer Statistics. J. Am. Acad. Dermatol. 2019, 80, 1154–1156. [Google Scholar] [CrossRef] [Green Version]
- Trinidad, C.M.; Torres-Cabala, C.A.; Prieto, V.G.; Aung, P.P. Update on eighth edition American Joint Committee on Cancer classification for Merkel cell carcinoma and histopathological parameters that determine prognosis. J. Clin. Pathol. 2019, 72, 337–340. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel Cell Carcinoma in the Age of Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernández-Figueras, M.T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Dereure, O.; Kluger, N.; Moles, J.; Guillot, B.; Segondy, M. Merkel cell polyomavirus DNA detection in lesional and nonlesional skin from patients with Merkel cell carcinoma or other skin diseases. Br. J. Dermatol. 2010, 162, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Kluger, N.; Dereure, O.; Mercier, G.; Molès, J.-P.; Guillot, B.; Segondy, M. Merkel cell polyomavirus in cutaneous swabs. Emerg. Infect. Dis. 2010, 16, 685. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Courgnaud, V.; Champeau, W.; Segondy, M. Detection of Merkel cell polyomavirus on environmental surfaces. J. Med. Virol. 2011, 83, 1435–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyo, M.; Guerrero-Preston, R.; Brait, M.; Hoque, M.O.; Chuang, A.; Kim, M.S.; Sharma, R.; Liégeois, N.J.; Koch, W.M.; Califano, J.A.; et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int. J. Cancer 2010, 126, 2991–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, U.; Mauch, C.; Kreuter, A.; Krieg, T.; Pfister, H. Merkel cell polyomavirus DNA in persons without merkel cell carcinoma. Emerg. Infect. Dis. 2009, 15, 1496. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Sáenz Robles, M.T.; Pipas, J.M. Large T Antigens of Polyomaviruses: Amazing Molecular Machines. Annu. Rev. Microbiol. 2012, 66, 213–236. [Google Scholar] [CrossRef]
- Campbell, K.S.; Auger, K.R.; Hemmings, B.A.; Roberts, T.M.; Pallas, D.C. Identification of regions in polyomavirus middle T and small t antigens important for association with protein phosphatase 2A. J. Virol. 1995, 69, 3721–3728. [Google Scholar] [CrossRef] [Green Version]
- Mullane Karen, P.; Ratnofsky, M.; Culleré, X.; Schaffhausen, B. Signaling from Polyomavirus Middle T and Small T Defines Different Roles for Protein Phosphatase 2A. Mol. Cell. Biol. 1998, 18, 7556–7564. [Google Scholar] [CrossRef] [Green Version]
- Kwun Hyun, J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore Patrick, S.; Chang, Y. The Minimum Replication Origin of Merkel Cell Polyomavirus Has a Unique Large T-Antigen Loading Architecture and Requires Small T-Antigen Expression for Optimal Replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Wang, R.; Nakamaru-Ogiso, E.; Knight, S.A.; Buck, C.B.; You, J. The oncogenic small tumor antigen of Merkel cell polyomavirus is an iron-sulfur cluster protein that enhances viral DNA replication. J. Virol. 2016, 90, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Small T Antigen Controls Viral Replication and Oncoprotein Expression by Targeting the Cellular Ubiquitin Ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerrahn, J.; Knippschild, U.; Winkler, T.; Deppert, W. Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J. 1993, 12, 4739–4746. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, S.K.; Ona-Vu, K.; Sullivan, E.M.; Dong, T.K.; Hsu, S.-W.; Oh, D.H. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int. J. Cancer 2012, 131, 1818–1827. [Google Scholar] [CrossRef]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. 2013, 110, 12744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Bayer, N.J.; Januliene, D.; Zocher, G.; Stehle, T.; Moeller, A.; Blaum, B.S. Structure of merkel cell polyomavirus capsid and interaction with its glycosaminoglycan attachment receptor. J. Virol. 2020, 94, e01664-19. [Google Scholar] [CrossRef]
- Forstová, J.; Krauzewicz, N.; Wallace, S.; Street, A.J.; Dilworth, S.M.; Beard, S.; Griffin, B.E. Cooperation of structural proteins during late events in the life cycle of polyomavirus. J. Virol. 1993, 67, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of Human Seroresponsiveness to Merkel Cell Polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [Green Version]
- Theiss, J.M.; Günther, T.; Alawi, M.; Neumann, F.; Tessmer, U.; Fischer, N.; Grundhoff, A. A Comprehensive Analysis of Replicating Merkel Cell Polyomavirus Genomes Delineates the Viral Transcription Program and Suggests a Role for mcv-miR-M1 in Episomal Persistence. PLoS Pathog. 2015, 11, e1004974. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Dürst, M.; Gissmann, L.; Ikenberg, H.; Zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses Are Chronically Shed from Human Skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialasiewicz, S.; Lambert, S.B.; Whiley, D.M.; Nissen, M.D.; Sloots, T.P. Merkel cell polyomavirus DNA in respiratory specimens from children and adults. Emerg. Infect. Dis. 2009, 15, 492. [Google Scholar] [CrossRef]
- Husseiny, M.I.; Anastasi, B.; Singer, J.; Lacey, S.F. A comparative study of Merkel cell, BK and JC polyomavirus infections in renal transplant recipients and healthy subjects. J. Clin. Virol. 2010, 49, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Bofill-Mas, S.; Rodriguez-Manzano, J.; Calgua, B.; Carratala, A.; Girones, R. Newly described human polyomaviruses Merkel Cell, KI and WU are present in urban sewage and may represent potential environmental contaminants. Virol. J. 2010, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Boulais, N.; Misery, L. Merkel cells. J. Am. Acad. Dermatol. 2007, 57, 147–165. [Google Scholar] [CrossRef]
- Moll, I.; Zieger, W.; Schmelz, M. Proliferative merkel cells were not detected in human skin. Arch. Dermatol. Res. 1996, 288, 184. [Google Scholar] [CrossRef]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, Gene Expression and Particle Production by a Consensus Merkel Cell Polyomavirus (MCPyV) Genome. PLoS ONE 2011, 6, e29112. [Google Scholar] [CrossRef] [Green Version]
- Schowalter, R.M.; Reinhold, W.C.; Buck, C.B. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS ONE 2012, 7, e42181. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Zhou, H.; Shuda, M.; Stolz, D.B.; Rapchak, K.; Moore, P.S.; Chang, Y. Replication Kinetics for a Reporter Merkel Cell Polyomavirus. Viruses 2022, 14, 473. [Google Scholar] [CrossRef] [PubMed]
- Siebels, S.; Czech-Sioli, M.; Spohn, M.; Schmidt, C.; Theiss, J.; Indenbirken, D.; Günther, T.; Grundhoff, A.; Fischer, N. Merkel Cell Polyomavirus DNA Replication Induces Senescence in Human Dermal Fibroblasts in a Kap1/Trim28-Dependent Manner. mBio 2020, 11, e00142-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.D.; Fullen, D.R.; Lowe, L.; Uherova, P.; Schnitzer, B.; Valdez, R. CD117 (KIT receptor) expression in Merkel cell carcinoma. Am. J. Dermatopathol. 2002, 24, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Deichmann, M.; Kurzen, H.; Egner, U.; Altevogt, P.; Hartschuh, W. Adhesion molecules CD171 (L1CAM) and CD24 are expressed by primary neuroendocrine carcinomas of the skin (Merkel cell carcinomas). J. Cutan. Pathol. 2003, 30, 363–368. [Google Scholar] [CrossRef]
- Calder, K.B.; Smoller, B.R. New insights into merkel cell carcinoma. Adv. Anat. Pathol. 2010, 17, 155–161. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Sunshine, J.; Jahchan, N.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Szeder, V.; Grim, M.; Halata, Z.; Sieber-Blum, M. Neural crest origin of mammalian Merkel cells. Dev. Biol. 2003, 253, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Breathnach, A.; Robins, J. Ultrastructural observations on Merkel cells in human foetal skin. J. Anat. 1970, 106, 411. [Google Scholar]
- Breathnach, A.S. Development and differentiation of dermal cells in man. J. Investig. Dermatol. 1978, 71, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. The ultrastructure of the skin of human embryos. X. Merkel tactile cells in the finger and nail. J. Anat. 1972, 111, 99–120. [Google Scholar] [PubMed]
- Perdigoto, C.N.; Bardot, E.S.; Valdes, V.J.; Santoriello, F.J.; Ezhkova, E. Embryonic maturation of epidermal Merkel cells is controlled by a redundant transcription factor network. Development 2014, 141, 4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdigoto, C.N.; Dauber, K.L.; Bar, C.; Tsai, P.-C.; Valdes, V.J.; Cohen, I.; Santoriello, F.J.; Zhao, D.; Zheng, D.; Hsu, Y.-C.; et al. Polycomb-Mediated Repression and Sonic Hedgehog Signaling Interact to Regulate Merkel Cell Specification during Skin Development. PLOS Genet. 2016, 12, e1006151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Keymeulen, A.; Mascre, G.; Youseff, K.K.; Harel, I.; Michaux, C.; De Geest, N.; Szpalski, C.; Achouri, Y.; Bloch, W.; Hassan, B.A.; et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J. Cell Biol. 2009, 187, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Bardot, E.S.; Valdes, V.J.; Zhang, J.; Perdigoto, C.N.; Nicolis, S.; Hearn, S.A.; Silva, J.M.; Ezhkova, E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 2013, 32, 1990–2000. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Thoresen, D.T.; Miao, L.; Williams, J.S.; Wang, C.; Atit, R.P.; Wong, S.Y.; Brownell, I. A Cascade of Wnt, Eda, and Shh Signaling Is Essential for Touch Dome Merkel Cell Development. PLoS Genet. 2016, 12, e1006150. [Google Scholar] [CrossRef] [Green Version]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.-M.; Berthon, P. Polyomavirus-positive Merkel cell carcinoma derived from a trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef]
- Gravemeyer, J.; Spassova, I.; Verhaegen, M.E.; Dlugosz, A.A.; Hoffmann, D.; Lange, A.; Becker, J.C. DNA-methylation patterns imply a common cellular origin of virus-and UV-associated Merkel cell carcinoma. Oncogene 2022, 41, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.M. Primary neuroendocrine (Merkel cell) carcinoma of the skin: Morphologic diversity and implications thereof. Hum. Pathol. 2001, 32, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, A.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolhe, R.; Reid, M.D.; Lee, J.R.; Cohen, C.; Ramalingam, P. Immunohistochemical expression of PAX5 and TdT by Merkel cell carcinoma and pulmonary small cell carcinoma: A potential diagnostic pitfall but useful discriminatory marker. Int. J. Clin. Exp. Pathol. 2013, 6, 142. [Google Scholar]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef] [Green Version]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; Schrama, D.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A. Antibodies to Merkel Cell Polyomavirus T Antigen Oncoproteins Reflect Tumor Burden in Merkel Cell Carcinoma Patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel Cell Polyomavirus-Infected Merkel Cell Carcinoma Cells Require Expression of Viral T Antigens. J. Virol. 2010, 84, 7064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino, P.J.; Robirds, D.H.; Tripp, S.R.; Pfeifer, J.D.; Abel, H.J.; Duncavage, E.J. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod. Pathol. 2014, 27, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [Green Version]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel Cell Polyomavirus Infection, Large T Antigen, Retinoblastoma Protein and Outcome in Merkel Cell Carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef] [Green Version]
- Richards, K.F.; Guastafierro, A.; Shuda, M.; Toptan, T.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J. Gen. Virol. 2015, 96, 3532. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Cheng, J.; Ward-Shaw, E.; Dick, F.A.; DeCaprio, J.A.; Lambert, P.F. Merkel cell polyomavirus large T antigen binding to pRb promotes skin hyperplasia and tumor development. PLoS Pathog. 2022, 18, e1010551. [Google Scholar] [CrossRef]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: A molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwun, H.J.; Shuda, M.; Camacho, C.J.; Gamper, A.M.; Thant, M.; Chang, Y.; Moore, P.S.; Imperiale, M.J. Restricted Protein Phosphatase 2A Targeting by Merkel Cell Polyomavirus Small T Antigen. J. Virol. 2015, 89, 4191–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel Cell Polyomavirus Small T Antigen Is Oncogenic in Transgenic Mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.H.; Simonette, R.A.; Hsiao, T.; Doan, H.Q.; Rady, P.L.; Tyring, S.K. Cutaneous human polyomavirus small T antigens and 4E-BP1 targeting. Intervirology 2015, 58, 382–385. [Google Scholar] [CrossRef]
- Velásquez, C.; Cheng, E.; Shuda, M.; Lee-Oesterreich, P.J.; Pogge von Strandmann, L.; Gritsenko, M.A.; Jacobs, J.M.; Moore, P.S.; Chang, Y. Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proc. Natl. Acad. Sci. 2016, 113, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Leiendecker, L.; Jung, P.S.; Krecioch, I.; Neumann, T.; Schleiffer, A.; Mechtler, K.; Wiesner, T.; Obenauf, A.C. LSD1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol. Med. 2020, 12, e12525. [Google Scholar] [CrossRef]
- Gupta, P.; Shahzad, N.; Harold, A.; Shuda, M.; Venuti, A.; Romero-Medina, M.C.; Pacini, L.; Brault, L.; Robitaille, A.; Taverniti, V. Merkel cell polyomavirus downregulates N-myc downstream-regulated gene 1, leading to cellular proliferation and migration. J. Virol. 2020, 94, e00899-19. [Google Scholar] [CrossRef]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Merkel cell polyomavirus in Merkel cell carcinogenesis: Small T antigen-mediates c-Jun phosphorylation. Virus Genes 2016, 52, 397–399. [Google Scholar] [CrossRef]
- Berrios, C.; Padi, M.; Keibler, M.A.; Park, D.E.; Molla, V.; Cheng, J.; Lee, S.M.; Stephanopoulos, G.; Quackenbush, J.; DeCaprio, J.A. Merkel cell polyomavirus small T antigen promotes pro-glycolytic metabolic perturbations required for transformation. PLoS Pathog. 2016, 12, e1006020. [Google Scholar] [CrossRef] [Green Version]
- Nwogu, N.; Boyne, J.R.; Dobson, S.J.; Poterlowicz, K.; Blair, G.E.; Macdonald, A.; Mankouri, J.; Whitehouse, A. Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PLoS Pathog. 2018, 14, e1007276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Jia, Y.; Shen, S.; Kim, J.; Wang, X.; Lee, E.; Brownell, I.; Cho-Vega, J.H.; Lewis, C.; Homsi, J. Merkel cell polyomavirus small T antigen activates noncanonical NF-κB signaling to promote tumorigenesis. Mol. Cancer Res. 2020, 18, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Pallas, D.C.; Shahrik, L.K.; Martin, B.L.; Jaspers, S.; Miller, T.B.; Brautigan, D.L.; Roberts, T.M. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 1990, 60, 167–176. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Samimi, M.; Hesbacher, S.; Berthon, P.; Wobser, M.; Sallot, A.; Sarma, B.; Schweinitzer, S.; Gandon, T.; Destrieux, C.; et al. Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers 2020, 12, 1989. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Harms, P.W.; Van Goor, J.J.; Arche, J.; Patrick, M.T.; Wilbert, D.; Zabawa, H.; Grachtchouk, M.; Liu, C.-J.; Hu, K.; et al. Direct cellular reprogramming enables development of viral T antigen–driven Merkel cell carcinoma in mice. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. 1992, 89, 12180–12184. [Google Scholar] [CrossRef] [Green Version]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Rose, R.C.; Bonnez, W.; Reichman, R.C.; Garcea, R.L. Expression of human papillomavirus type 11 L1 protein in insect cells: In vivo and in vitro assembly of viruslike particles. J. Virol. 1993, 67, 1936–1944. [Google Scholar] [CrossRef] [Green Version]
- Schiller, J.T.; Hidesheim, A. Developing HPV virus-like particle vaccines to prevent cervical cancer: A progress report. J. Clin. Virol. 2000, 19, 67–74. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D. Production of Papillomavirus-Based Gene Transfer Vectors. Curr. Protoc. Cell Biol. 2007, 37, 26.21.21–26.21.19. [Google Scholar] [CrossRef] [PubMed]
- Gilson, T.D.; Gibson, R.T.; Androphy, E.J. Optimization of human papillomavirus-based pseudovirus techniques for efficient gene transfer. Sci. Rep. 2020, 10, 15517. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 2008, 82, 12565–12568. [Google Scholar] [CrossRef] [Green Version]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [Green Version]
- Day, P.M.; Weisberg, A.S.; Thompson, C.D.; Hughes, M.M.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Human papillomavirus 16 capsids mediate nuclear entry during infection. J. Virol. 2019, 93, e00454-19. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-S.; Lambert, P.F. Use of an in vivo animal model for assessing the role of integrin α6Β4 and Syndecan-1 in early steps in papillomavirus infection. Virology 2012, 433, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Selinka, H.-C.; Giroglou, T.; Sapp, M. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 2002, 299, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and Sialylated Glycans Sequentially Facilitate Merkel Cell Polyomavirus Infectious Entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef] [Green Version]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef] [Green Version]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different Heparan Sulfate Proteoglycans Serve asCellular Receptors for HumanPapillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Hoffmann, B.L.; Schlosser, S.J.; Ivarie, C.A.R.; Meisner, L.F.; O’Connor, S.L.; Sattler, C.A. Normal Growth and Differentiation in a Spontaneously Immortalized Near-Diploid Human Keratinocyte Cell Line, NIKS. J. Investig. Dermatol. 2000, 114, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Sattler, C.A.; Lambert, P.F. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 1999, 262, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The Human Papillomavirus Type 16 E7 Oncogene Is Required for the Productive Stage of the Viral Life Cycle. J. Viro. 2000, 74, 6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using Organotypic Epithelial Tissue Culture to Study the Human Papillomavirus Life Cycle. Curr. Protoc. Microbiol. 2016, 41, 14b.18.11–14b.18.19. [Google Scholar] [CrossRef]
- Loke, A.S.W.; Longley, B.J.; Lambert, P.F.; Spurgeon, M.E. A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus-Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin. Viruses 2021, 13, 138. [Google Scholar] [CrossRef]
- Temblador, A.; Topalis, D.; van den Oord, J.; Andrei, G.; Snoeck, R. Organotypic Epithelial Raft Cultures as a Three-Dimensional In Vitro Model of Merkel Cell Carcinoma. Cancers 2022, 14, 1091. [Google Scholar] [CrossRef]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S.; et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab Invest. 1987, 56, 302–312. [Google Scholar]
- Guastafierro, A.; Feng, H.; Thant, M.; Kirkwood, J.M.; Chang, Y.; Moore, P.S.; Shuda, M. Characterization of an early passage Merkel cell polyomavirus-positive Merkel cell carcinoma cell line, MS-1, and its growth in NOD scid gamma mice. J. Virol. Methods 2013, 187, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-Positive Merkel Cell Carcinoma Cell Line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Murphy, J.B.; Rous, P. The behavior of chicken sarcoma implanted in the developing embryo. J. Exp. Med. 1912, 15, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.; Yatsyna, A.; Charbonneau, M.; Brochu-Gaudreau, K.; Perreault, A.; Jeldres, C.; McDonald, P.P.; Dubois, C.M. The Chicken Chorioallantoic Membrane Tumor Assay as a Relevant In Vivo Model to Study the Impact of Hypoxia on Tumor Progression and Metastasis. Cancers 2021, 13, 1093. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.T.; Shahin, S.A.; Croissant, J.; Fatieiev, Y.; Matsumoto, K.; Le-Hoang Doan, T.; Yik, T.; Simargi, S.; Conteras, A.; Ratliff, L.; et al. Chick chorioallantoic membrane assay as an in vivo model to study the effect of nanoparticle-based anticancer drugs in ovarian cancer. Sci. Rep. 2018, 8, 8524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, P.Y.; Koh, A.P.F.; Antony, J.; Huang, R.Y.J. Applications of the Chick Chorioallantoic Membrane as an Alternative Model for Cancer Studies. Cells Tissues Organs 2022, 211, 222–237. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM). A multifaceted experimental model. Mech. Dev. 2016, 141, 70–77. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod. Toxicol. 2017, 70, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.K.; Krump, C.; Bernhart, E.; Becker, J.C.; Sattler, W.; Ghaffari-Tabrizi-Wizsy, N. A short-term in vivo model for Merkel Cell Carcinoma. Exp. Dermatol. 2018, 27, 684–687. [Google Scholar] [CrossRef] [Green Version]
- Knips, J.; Czech-Sioli, M.; Spohn, M.; Heiland, M.; Moll, I.; Grundhoff, A.; Schumacher, U.; Fischer, N. Spontaneous lung metastasis formation of human Merkel cell carcinoma cell lines transplanted into scid mice. Int. J. Cancer 2017, 141, 160–171. [Google Scholar] [CrossRef]
- Krasagakis, K.; Almond-Roesler, B.; Geilen, C.; Fimmel, S.; Krengel, S.; Chatzaki, E.; Gravanis, A.; Orfanos, C.E. Growth and characterization of a cell line from a human primary neuroendocrine carcinoma of the skin (Merkel cell carcinoma) in culture and as xenograft. J. Cell. Physiol. 2001, 187, 386–391. [Google Scholar] [CrossRef]
- Fang, B.; Kannan, A.; Zhao, S.; Nguyen, Q.H.; Ejadi, S.; Yamamoto, M.; Camilo Barreto, J.; Zhao, H.; Gao, L. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci. Rep. 2020, 10, 8867. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT Pathway in Merkel Cell Carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, Y.; Mao, J.H.; Brown, K.; Girardi, M.; Balmain, A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature 2007, 445, 761–765. [Google Scholar] [CrossRef]
- Hennings, H.; Glick, A.B.; Lowry, D.T.; Krsmanovic, L.S.; Sly, L.M.; Yuspa, S.H. FVB/N mice: An inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis 1993, 14, 2353–2358. [Google Scholar] [CrossRef]
- Brake, T.; Connor, J.P.; Petereit, D.G.; Lambert, P.F. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: Identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003, 63, 8173–8180. [Google Scholar]
- Spurgeon, M.E.; Liem, A.; Buehler, D.; Cheng, J.; DeCaprio, J.A.; Lambert, P.F. The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin. Cancers 2021, 13, 222. [Google Scholar] [CrossRef]
- Balsitis, S.; Dick, F.; Lee, D.; Farrell, L.; Hyde, R.K.; Griep Anne, E.; Dyson, N.; Lambert Paul, F. Examination of the pRb-Dependent and pRb-Independent Functions of E7 In Vivo. J. Virol. 2005, 79, 11392–11402. [Google Scholar] [CrossRef] [Green Version]
- Balsitis, S.; Dick, F.; Dyson, N.; Lambert, P.F. Critical Roles for Non-pRb Targets of Human Papillomavirus Type 16 E7 in Cervical Carcinogenesis. Cancer Res. 2006, 66, 9393–9400. [Google Scholar] [CrossRef] [Green Version]
- Dick Frederick, A.; Sailhamer, E.; Dyson Nicholas, J. Mutagenesis of the pRB Pocket Reveals that Cell Cycle Arrest Functions Are Separable from Binding to Viral Oncoproteins. Mol. Cel. Biol. 2000, 20, 3715–3727. [Google Scholar] [CrossRef]
- Dick Frederick, A.; Dyson Nicholas, J. Three Regions of the pRB Pocket Domain Affect Its Inactivation by Human Papillomavirus E7 Proteins. J. Virol. 2002, 76, 6224–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac Christian, E.; Francis Sarah, M.; Martens Alison, L.; Julian Lisa, M.; Seifried Laurie, A.; Erdmann, N.; Binné Ulrich, K.; Harrington, L.; Sicinski, P.; Bérubé Nathalie, G.; et al. The Retinoblastoma Protein Regulates Pericentric Heterochromatin. Mol. Cell. Biol. 2006, 26, 3659–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.B.; Cohen, I.; Kumar, V.; Xu, Z.; Bar, C.; Dauber-Decker, K.L.; Tsai, P.C.; Marangoni, P.; Klein, O.D.; Hsu, Y.C.; et al. FGF signalling controls the specification of hair placode-derived SOX9 positive progenitors to Merkel cells. Nat. Commun. 2018, 9, 2333. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loke, A.S.W.; Lambert, P.F.; Spurgeon, M.E. Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC. Viruses 2022, 14, 2204. https://doi.org/10.3390/v14102204
Loke ASW, Lambert PF, Spurgeon ME. Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC. Viruses. 2022; 14(10):2204. https://doi.org/10.3390/v14102204
Chicago/Turabian StyleLoke, Amanda S. W., Paul F. Lambert, and Megan E. Spurgeon. 2022. "Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC" Viruses 14, no. 10: 2204. https://doi.org/10.3390/v14102204
APA StyleLoke, A. S. W., Lambert, P. F., & Spurgeon, M. E. (2022). Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC. Viruses, 14(10), 2204. https://doi.org/10.3390/v14102204