
Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Epidemiology
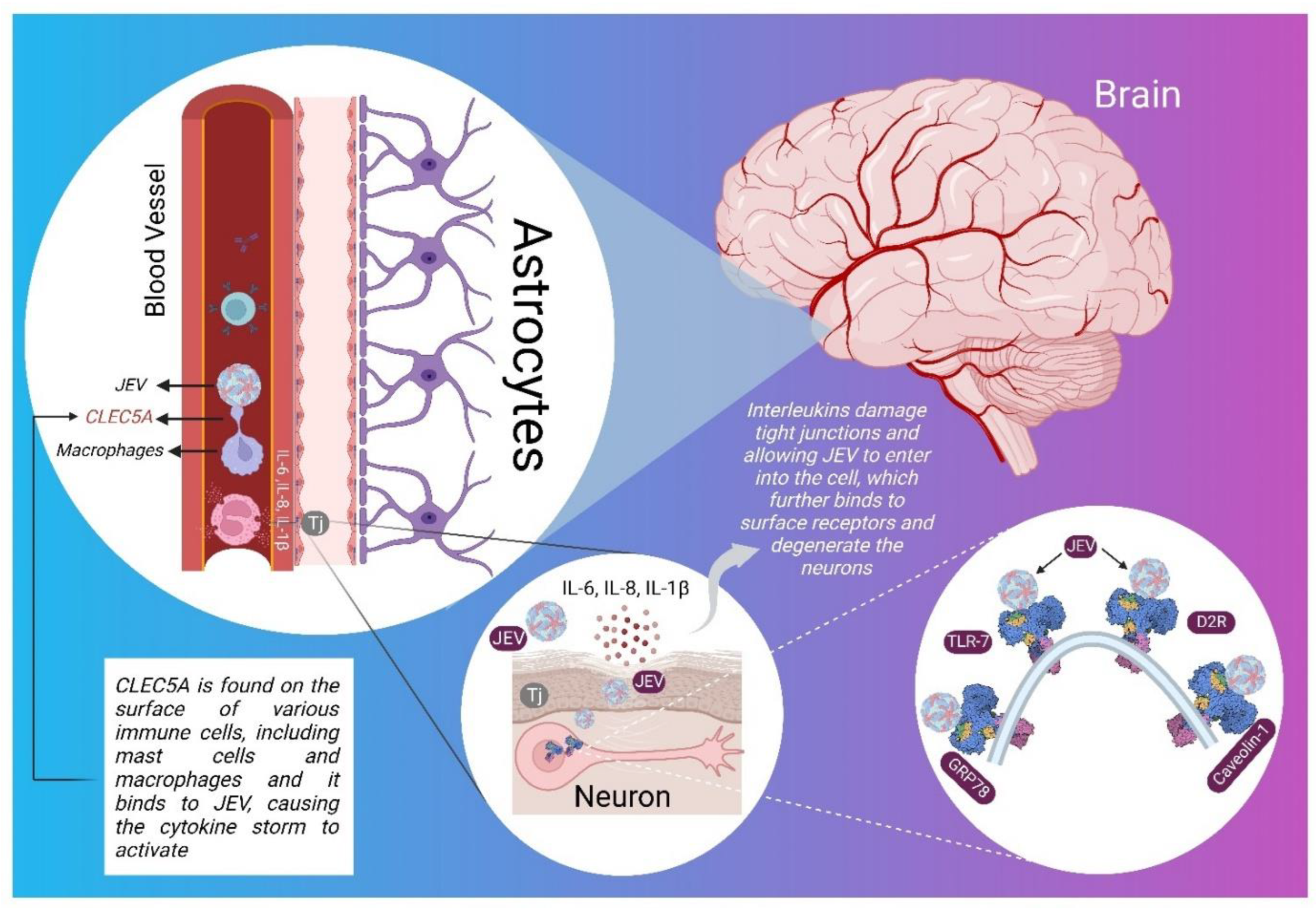
3. Pathophysiology of JE in CNS
4. Overview of the JEV Genome
5. Molecular Targets Associated with JEV Infection
5.1. CLEC5A
5.2. GRP78
5.3. Caveolin-1
5.4. D2R
5.5. TLRs
5.6. Src Protein
6. Autoimmunity in JEV Infection
7. Roles of Different ILs
7.1. IL-6
7.2. IL-8
7.3. IL-10
7.4. Other ILs
8. Therapeutic Insights: Possibilities against JEV Infection in CNS
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mulvey, P.; Duong, V.; Boyer, S.; Burgess, G.; Williams, D.T.; Dussart, P.; Horwood, P.F. The Ecology and Evolution of Japanese Encephalitis Virus. Pathogens 2021, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhao, Z. Vascular inflammation in the central nervous system. Neural. Regen. Res. 2022, 17, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.; Ragupathy, S.; Borchard, G. Target specific tight junction modulators. Adv. Drug. Deliv. Rev. 2021, 171, 266–288. [Google Scholar] [CrossRef] [PubMed]
- Pinapati, K.K.; Tandon, R.; Tripathi, P.; Srivastava, N. Recent advances to overcome the burden of Japanese encephalitis: A zoonotic infection with problematic early detection. Rev. Med. Virol. 2022, e2383. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Yu, L.; Cao, S.; Wang, K.; Yuan, J.; Wang, C.; Wang, K.; Cui, M.; Fu, Z.F. Viral Infection of the Central Nervous System and Neuroinflammation Precede Blood-Brain Barrier Disruption during Japanese Encephalitis Virus Infection. J. Virol. 2015, 89, 5602–5614. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, M.J.; Turtle, L.; Solomon, T. Japanese encephalitis virus infection. Handb. Clin. Neurol. 2014, 123, 561–576. [Google Scholar]
- Lindquist, L. Recent and historical trends in the epidemiology of Japanese encephalitis and its implication for risk assessment in travellers. J. Travel. Med. 2018, 25 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Wangchuk, S.; Tamang, T.D.; Darnal, J.B.; Pelden, S.; Lhazeen, K.; Mynak, M.L.; Letson, G.W.; Khare, S.; Leader, B.T.; Marfin, A.A.; et al. Japanese Encephalitis Virus as Cause of Acute Encephalitis, Bhutan. Emerg. Infect. Dis. 2020, 26, 2239–2242. [Google Scholar] [CrossRef]
- Moore, S.M. The current burden of Japanese encephalitis and the estimated impacts of vaccination: Combining estimates of the spatial distribution and transmission intensity of a zoonotic pathogen. PLoS Negl. Trop. Dis. 2021, 15, e0009385. [Google Scholar] [CrossRef]
- Le Flohic, G.; Porphyre, V.; Barbazan, P.; Gonzalez, J.P. Review of climate, landscape, and viral genetics as drivers of the Japanese encephalitis virus ecology. PLoS Negl. Trop. Dis. 2013, 7, e2208. [Google Scholar] [CrossRef] [Green Version]
- Hegde, N.R.; Gore, M.M. Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccin. Immunother. 2017, 13, 1320–1337. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Sharma, S.; Hans, C. Japanese Encephalitis and Its Epidemiology. J. Infect. Dis. Ther. 2015, 3, 243. [Google Scholar] [CrossRef]
- Wang, H.; Liang, G. Epidemiology of Japanese encephalitis: Past, present, and future prospects. Ther. Clin. Risk. Manag. 2015, 11, 435–448. [Google Scholar] [PubMed] [Green Version]
- Kumar, R. Viral encephalitis of public health significance in India: Current status. Indian J. Pediatr. 1999, 66, 73–83. [Google Scholar] [CrossRef]
- Mathur, A.; Chaturvedi, U.C.; Tandon, H.O.; Agarwal, A.K.; Mathur, G.P.; Nag, D.; Prasad, A.; Mittal, V.P. Japanese encephalitis epidemic in Uttar Pradesh, India during 1978. Indian J. Med. Res. 1982, 75, 161–169. [Google Scholar] [PubMed]
- Kumar, R.; Tripathi, P.; Rizvi, A. Effectiveness of one dose of SA 14-14-2 vaccine against Japanese encephalitis. N. Engl. J. Med. 2009, 360, 1465–1466. [Google Scholar] [CrossRef]
- Li, J.; Chen, H.; Wu, N.; Fan, D.; Liang, G.; Gao, N.; An, J. Characterization of immune responses induced by inactivated, live attenuated and DNA vaccines against Japanese encephalitis virus in mice. Vaccine 2013, 31, 4136–4142. [Google Scholar] [CrossRef]
- Vashishtha, V.M.; Kalra, A.; Bose, A.; Choudhury, P.; Yewale, V.N.; Bansal, C.P.; Gupta, S.G. Indian Academy of Pediatrics (IAP) recommended immunization schedule for children aged 0 through 18 years—India, 2013 and updates on immunization. Indian Pediatr. 2013, 50, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Dumre, S.P.; Shakya, G.; Na-Bangchang, K.; Eursitthichai, V.; Rudi Grams, H.; Upreti, S.R.; Ghimire, P.; Khagendra, K.C.; Nisalak, A.; Gibbons, R.V.; et al. Dengue virus and Japanese encephalitis virus epidemiological shifts in Nepal: A case of opposing trends. Am. J. Trop. Med. Hyg. 2013, 88, 677–680. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Kawado, M.; Murakami, Y.; Izumida, M.; Ohta, A.; Tada, Y.; Shigematsu, M.; Yasui, Y.; Taniguchi, K.; Nagai, M. Epidemics of vector-borne diseases observed in infectious disease surveillance in Japan, 2000–2005. J. Epidemiol. 2007, 17 (Suppl. 1), S48–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Matsunaga, Y.; Takasaki, T.; Tanaka-Taya, K.; Taniguchi, K.; Okabe, N.; Kurane, I. Vaccine Preventable Diseases Surveillance Program of Japan. Japanese encephalitis: Surveillance and elimination effort in Japan from 1982 to 2004. Jpn. J. Infect. Dis. 2008, 61, 333–338. [Google Scholar] [PubMed]
- Ramli, N.S.; Ismail, N.M.; Zaini, N.; Hayati, F.; Jeffree, M.S.; Abdul Rahim, S.S.S.; Hassan, M.R. Seroepidemiological Studies on Japanese Encephalitis: A Systematic Review. Oman Med. J. 2022, 37, e366. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.N.; Ritchie, S.A.; Phillips, D.A.; Lee, J.M.; Hills, S.L.; van den Hurk, A.F.; Pyke, A.T.; Johansen, C.A.; Mackenzie, J.S. Japanese encephalitis in north Queensland, Australia, 1998. Med. J. Aust. 1999, 170, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.M.; Thao, T.T.N.; Duy, N.M.; Nhat, T.M.; Clapham, H. Estimates of the global burden of Japanese encephalitis and the impact of vaccination from 2000–2015. elife 2020, 9, e51027. [Google Scholar] [CrossRef]
- Xufang, Y.; Huanyu, W.; Shihong, F.; Xiaoyan, G.; Shuye, Z.; Chunting, L.; Minghua, L.; Yougang, Z.; Guodong, L. Etiological spectrum of clinically diagnosed Japanese encephalitis cases reported in Guizhou Province, China, in 2006. J. Clin. Microbiol. 2010, 48, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Wang, H.; Yang, J.; Luo, H.; Li, Y.; Hadler, S.C.; Sandhu, H.S.; Fischer, M.; Jiang, Y.; Zhang, Z.; et al. Acute Meningitis and Encephalitis Syndrome (AMES) Study Group. Japanese encephalitis disease burden and clinical features of Japanese encephalitis in four cities in the People’s Republic of China. Am. J. Trop. Med. Hyg. 2010, 83, 766–773. [Google Scholar] [CrossRef]
- Kari, K.; Liu, W.; Gautama, K.; Mammen, M.P., Jr.; Clemens, J.D.; Nisalak, A.; Subrata, K.; Kim, H.K.; Xu, Z.Y. A hospital-based surveillance for Japanese encephalitis in Bali, Indonesia. BMC Med. 2006, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Touch, S.; Hills, S.; Sokhal, B.; Samnang, C.; Sovann, L.; Khieu, V.; Soeung, S.C.; Toda, K.; Robinson, J.; Grundy, J. Epidemiology and burden of disease from Japanese encephalitis in Cambodia: Results from two years of sentinel surveillance. Trop. Med. Int. Health. 2009, 14, 1365–1373. [Google Scholar] [CrossRef]
- Baig, S.; Fox, K.K.; Jee, Y.; O’Connor, P.; Hombach, J.; Wang, S.A.; Hyde, T.; Fischer, M.; Hills, S.L. Japanese encephalitis surveillance and immunization—Asia and the Western Pacific, 2012. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 658. [Google Scholar]
- Zimmerman, M.D.; Scott, R.M.; Vaughn, D.W.; Rajbhandari, S.; Nisalak, A.; Shrestha, M.P. Short report: An outbreak of Japanese encephalitis in Kathmandu, Nepal. Am. J. Trop. Med. Hyg. 1997, 57, 283–284. [Google Scholar] [CrossRef]
- Partridge, J.; Ghimire, P.; Sedai, T.; Bista, M.B.; Banerjee, M. Endemic Japanese encephalitis in the Kathmandu valley, Nepal. Am. J. Trop. Med. Hyg. 2007, 77, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Wierzba, T.F.; Ghimire, P.; Malla, S.; Banerjee, M.K.; Shrestha, S.; Khanal, B.; Sedai, T.R.; Gibbons, R.V. Laboratory-based Japanese encephalitis surveillance in Nepal and the implications for a national immunization strategy. Am. J. Trop. Med. Hyg. 2008, 78, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.J.; Supawat, K.; Campbell, A.P.; Anantapreecha, S.; Liamsuwan, S.; Tunlayadechanont, S.; Visudtibhan, A.; Lupthikulthum, S.; Dhiravibulya, K.; Viriyavejakul, A.; et al. Japanese encephalitis virus remains an important cause of encephalitis in Thailand. Int. J. Infect. Dis. 2010, 14, e888–e892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeewandara, C.; Gomes, L.; Paranavitane, S.A.; Tantirimudalige, M.; Panapitiya, S.S.; Jayewardene, A.; Fernando, S.; Fernando, R.H.; Prathapan, S.; Ogg, G.S.; et al. Change in Dengue and Japanese Encephalitis Seroprevalence Rates in Sri Lanka. PLoS ONE 2015, 10, e0144799. [Google Scholar] [CrossRef] [Green Version]
- Van den Hurk, A.F.; Pyke, A.T.; Mackenzie, J.S.; Hall-Mendelin, S.; Ritchie, S.A. Japanese Encephalitis Virus in Australia: From Known Known to Known Unknown. Trop. Med. Infect. Dis. 2019, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Paul, K.K.; Sazzad, H.M.S.; Rahman, M.; Sultana, S.; Hossain, M.J.; Ledermann, J.P.; Burns, P.; Friedman, M.S.; Flora, M.S.; Fischer, M.; et al. Hospital-based surveillance for Japanese encephalitis in Bangladesh, 2007-2016: Implications for introduction of immunization. Int. J. Infect. Dis. 2020, 99, 69–74. [Google Scholar] [CrossRef]
- Hu, X.T.; Li, Q.F.; Ma, C.; Zhao, Z.X.; He, L.F.; Tang, T.T.; Yu, W.; Owiti, P. Reduction patterns of Japanese encephalitis incidence following vaccine introduction into long-term expanded program on immunization in Yunnan Province, China. Infect. Dis. Poverty. 2019, 8, 102. [Google Scholar] [CrossRef]
- India Action Taken by Govt. of India towards Prevention & Control of AES/JE. National Center for Vector Borne Diseases Control (NCVBDC). Available online: https://nvbdcp.gov.in/index4.php?lang=1&level=0&linkid=475&lid=3762 (accessed on 21 August 2022).
- Garjito, T.A.; Widiarti; Anggraeni, Y.M.; Alfiah, S.; Tunggul Satoto, T.B.; Farchanny, A.; Samaan, G.; Afelt, A.; Manguin, S.; Frutos, R.; et al. Japanese encephalitis in Indonesia: An update on epidemiology and transmission ecology. Acta Trop. 2018, 187, 240–247. [Google Scholar] [CrossRef]
- Mayxay, M.; Douangdala, P.; Vilayhong, C.; Phommasone, K.; Chansamouth, V.; Vongsouvath, M.; Rattanavong, S.; Chang, K.; Sengvilaipaseuth, O.; Chanthongthip, A.; et al. Outcome of Japanese Encephalitis Virus (JEV) Infection in Pediatric and Adult Patients at Mahosot Hospital, Vientiane, Lao PDR. Am. J. Trop. Med. Hyg. 2020, 104, 567–575. [Google Scholar] [CrossRef]
- Kumar, K.; Arshad, S.S.; Selvarajah, G.T.; Abu, J.; Toung, O.P.; Abba, Y.; Yasmin, A.R.; Bande, F.; Sharma, R.; Ong, B.L. Japanese encephalitis in Malaysia: An overview and timeline. Acta Trop. 2018, 185, 219–229. [Google Scholar] [CrossRef]
- Hanson, J.P.; Taylor, C.T.; Richards, A.R.; Smith, I.L.; Boutlis, C.S. Japanese encephalitis acquired near Port Moresby: Implications for residents and travellers to Papua New Guinea. Med. J. Aust. 2004, 181, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Paul, W.S.; Moore, P.S.; Karabatsos, N.; Flood, S.P.; Yamada, S.; Jackson, T.; Tsai, T.F. Outbreak of Japanese encephalitis on the island of Saipan, 1990. J. Infect. Dis. 1993, 167, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.S.; Park, O.; Kong, I.S. Review of the Incidence of Japanese Encephalitis in Foreign-Born and Korean Nationals Living in the Republic of Korea, 2007–2016. Osong Public Health Res. Perspect. 2018, 9, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Japanese Encephalitis. Taiwan Centers for Disease Control. Available online: https://www.cdc.gov.tw/En/Category/ListContent/bg0g_VU_Ysrgkes_KRUDgQ?uaid=FCBms2B8k0PJx4io35AsOw (accessed on 21 August 2022).
- Chen, T.; He, X.; Zhang, P.; Yuan, Y.; Lang, X.; Yu, J.; Qin, Z.; Li, X.; Zhang, Q.; Zhu, L.; et al. Research advancements in the neurological presentation of flaviviruses. Rev. Med. Virol. 2019, 29, e2021. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Mustafá, Y.M.; Meuren, L.M.; Coelho, S.V.A.; de Arruda, L.B. Pathways Exploited by Flaviviruses to Counteract the Blood-Brain Barrier and Invade the Central Nervous System. Front. Microbiol. 2019, 10, 525. [Google Scholar] [CrossRef]
- Wang, K.; Wang, H.; Lou, W.; Ma, L.; Li, Y.; Zhang, N.; Wang, C.; Li, F.; Awais, M.; Cao, S.; et al. IP-10 Promotes Blood-Brain Barrier Damage by Inducing Tumor Necrosis Factor Alpha Production in Japanese Encephalitis. Front. Immunol. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Biswas, S.M.; Kar, S.; Singh, R.; Chakraborty, D.; Vipat, V.; Raut, C.G.; Mishra, A.C.; Gore, M.M.; Ghosh, D. Immunomodulatory cytokines determine the outcome of Japanese encephalitis virus infection in mice. J. Med. Virol. 2010, 82, 304–310. [Google Scholar] [CrossRef]
- Li, Y.; Ashraf, U.; Chen, Z.; Zhou, D.; Imran, M.; Ye, J.; Chen, H.; Cao, S. Genome-wide profiling of host-encoded circular RNAs highlights their potential role during the Japanese encephalitis virus-induced neuroinflammatory response. BMC Genom. 2020, 21, 409. [Google Scholar] [CrossRef]
- Warke, R.V.; Xhaja, K.; Martin, K.J.; Fournier, M.F.; Shaw, S.K.; Brizuela, N.; de Bosch, N.; Lapointe, D.; Ennis, F.A.; Rothman, A.L.; et al. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J. Virol. 2003, 77, 11822–11832. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.L.; Ng, M.L. Molecular mechanisms of West Nile virus pathogenesis in brain cell. Emerg. Infect. Dis. 2005, 11, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Venter, M.; Myers, T.G.; Wilson, M.A.; Kindt, T.J.; Paweska, J.T.; Burt, F.J.; Leman, P.A.; Swanepoel, R. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology 2005, 342, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Das, S.; Ghosh, S.; Basu, A. Tumor necrosis factor receptor-1-induced neuronal death by TRADD contributes to the pathogenesis of Japanese encephalitis. J. Neurochem. 2007, 103, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mehta, D.; Mishra, N.; Nayak, D.; Sunil, S. Role of Host-Mediated Post-Translational Modifications (PTMs) in RNA Virus Pathogenesis. Int. J. Mol. Sci. 2020, 22, 323. [Google Scholar] [CrossRef]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Jheng, J.R.; Ho, J.Y.; Horng, J.T. ER stress, autophagy, and RNA viruses. Front. Microbiol. 2014, 5, 388. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Yu, Y.; Deubel, V. Japanese encephalitis virus NS1′ protein depends on pseudoknot secondary structure and is cleaved by caspase during virus infection and cell apoptosis. Microbes Infect. 2012, 14, 930–940. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Gu, J.; Fan, Y.; Zhao, P.; Cao, R.; Chen, P. The A66G back mutation in NS2A of JEV SA14-14-2 strain contributes to production of NS1′ protein and the secreted NS1′ can be used for diagnostic biomarker for virulent virus infection. Infect Genet. Evol. 2015, 36, 116–125. [Google Scholar] [CrossRef]
- Ashraf, U.; Ding, Z.; Deng, S.; Ye, J.; Cao, S.; Chen, Z. Pathogenicity and virulence of Japanese encephalitis virus: Neuroinflammation and neuronal cell damage. Virulence. 2021, 12, 968–980. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, A.; Yadav, P.; Dubey, S.K.; Azhar, E.I.; Maitra, S.S.; Dwivedi, V.D. Molecular pathogenesis of Japanese encephalitis and possible therapeutic strategies. Arch. Virol. 2022, 167, 1739–1762. [Google Scholar] [CrossRef]
- Yun, S.I.; Choi, Y.J.; Song, B.H.; Lee, Y.M. 3′ cis-acting elements that contribute to the competence and efficiency of Japanese encephalitis virus genome replication: Functional importance of sequence duplications, deletions, and substitutions. J. Virol. 2009, 83, 7909–7930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, A.J.; Ward, M.J.; Brown, A.J.; Barrett, A.D. Phylogeography of Japanese encephalitis virus: Genotype is associated with climate. PLoS Negl. Trop. Dis. 2013, 7, e2411. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.T.; Chang, B.L.; Liang, J.J.; Tsai, H.J.; Lee, Y.L.; Lin, R.J.; Lin, Y.L. Japanese encephalitis virus nonstructural protein NS5 interacts with mitochondrial trifunctional protein and impairs fatty acid β-oxidation. PLoS Pathog. 2015, 11, e1004750. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Liu, R.S.; Wu, M.F.; Lin, Y.L.; Chen, S.Y.; Tan, D.T.; Chou, T.Y.; Tsai, I.S.; Li, L.; Hsieh, S.L. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog. 2012, 8, e1002655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.S.; Huang, T.F.; Hsieh, S.L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.; Shen, F.; Phillips, J.H.; McGeachy, M.J.; Cua, D.J.; Heyworth, P.G.; Pierce, R.H. Activation of MDL-1 (CLEC5A) on immature myeloid cells triggers lethal shock in mice. J. Clin. Invest. 2011, 121, 4446–4461. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Peláez, M.A.; Velandia-Romero, M.L.; Bastidas-Legarda, L.Y.; Beltrán, E.O.; Camacho-Ortega, S.J.; Castellanos, J.E. Dengue Virus Infection of Blood-Brain Barrier Cells: Consequences of Severe Disease. Front. Microbiol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Gilthorpe, J.; van der Maarel, J.R. MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fibrillization. PLoS ONE 2012, 7, e32953. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Quinones, Q.J.; de Ridder, G.G.; Pizzo, S.V. GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 2008, 23, v1409–v1416. [Google Scholar]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91, e02274-16. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.G.; Chen, Y.; Wang, X.; Zhao, P.; Zhu, Y.; Qi, Z. Ezrin is essential for the entry of Japanese encephalitis virus into the human brain microvascular endothelial cells. Emerg. Microbes Infect. 2020, 9, 1330–1341. [Google Scholar] [CrossRef]
- Fiala, G.J.; Minguet, S. Caveolin-1: The Unnoticed Player in TCR and BCR Signaling. Adv. Immunol. 2018, 137, 83–133. [Google Scholar] [PubMed]
- Xu, Q.; Cao, M.; Song, H.; Chen, S.; Qian, X.; Zhao, P.; Ren, H.; Tang, H.; Wang, Y.; Wei, Y.; et al. Caveolin-1-mediated Japanese encephalitis virus entry requires a two-step regulation of actin reorganization. Future Microbiol. 2016, 11, 1227–1248. [Google Scholar] [CrossRef]
- Simanjuntak, Y.; Liang, J.J.; Lee, Y.L.; Lin, Y.L. Japanese Encephalitis Virus Exploits Dopamine D2 Receptor-phospholipase C to Target Dopaminergic Human Neuronal Cells. Front. Microbiol. 2017, 8, 651. [Google Scholar] [CrossRef] [Green Version]
- Laureti, M.; Narayanan, D.; Rodriguez-Andres, J.; Fazakerley, J.K.; Kedzierski, L. Flavivirus Receptors: Diversity, Identity, and Cell Entry. Front. Immunol. 2018, 9, 2180. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18. [Google Scholar] [CrossRef]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Chae, H.D.; Choi, S.Y.; Kim, K.T. Transcriptional enhancement of tyrosine hydroxylase by prostaglandin E2 in SK-N-BE(2) C cells. Brain Res. Mol. Brain Res. 1996, 39, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Marcellino, D.; Kehr, J.; Agnati, L.F.; Fuxe, K. Increased affinity of dopamine for D(2) -like versus D(1) -like receptors. Relevance for volume transmission in interpreting PET findings. Synapse 2012, 66, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Simanjuntak, Y.; Liang, J.J.; Lee, Y.L.; Lin, Y.L. Repurposing of prochlorperazine for use against dengue virus infection. J. Infect. Dis. 2015, 211, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Koller, W.; Herbster, G.; Anderson, D.; Wack, R.; Gordon, J. Quinpirole hydrochloride, a potential anti-parkinsonism drug. Neuropharmacol. 1987, 26, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Trinquet, E.; Fink, M.; Bazin, H.; Grillet, F.; Maurin, F.; Bourrier, E.; Ansanay, H.; Leroy, C.; Michaud, A.; Durroux, T.; et al. D-myo-inositol 1-phosphate as a surrogate of D-myo-inositol 1,4,5-tris phosphate to monitor G protein-coupled receptor activation. Anal. Biochem. 2006, 358, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, A.; Das, S.; Ghosh, S.; Mishra, M.K.; Sharma, V.; Koli, P.; Sen, E.; Basu, A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia. 2007, 55, 483–496. [Google Scholar] [CrossRef]
- Awais, M.; Wang, K.; Lin, X.; Qian, W.; Zhang, N.; Wang, C.; Wang, K.; Zhao, L.; Fu, Z.F.; Cui, M. TLR7 Deficiency Leads to TLR8 Compensative Regulation of Immune Response against JEV in Mice. Front. Immunol. 2017, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Basu, A. Japanese encephalitis-a pathological and clinical perspective. PLoS Negl. Trop. Dis. 2009, 3, e437. [Google Scholar] [CrossRef] [Green Version]
- Bourne, N.; Scholle, F.; Silva, M.C.; Rossi, S.L.; Dewsbury, N.; Judy, B.; De Aguiar, J.B.; Leon, M.A.; Estes, D.M.; Fayzulin, R.; et al. Early production of type I interferon during West Nile virus infection: Role for lymphoid tissues in IRF3-independent interferon production. J. Virol. 2007, 81, 9100–9108. [Google Scholar] [CrossRef] [Green Version]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.K.; et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010, 6, e1000757. [Google Scholar] [CrossRef] [PubMed]
- Scholle, F.; Mason, P.W. West Nile virus replication interferes with both poly(I:C)-induced interferon gene transcription and response to interferon treatment. Virology 2005, 342, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M., Jr.; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef] [PubMed]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.W.; Choi, J.Y.; Uyangaa, E.; Kim, S.B.; Kim, J.H.; Kim, B.S.; Kim, K.; Eo, S.K. Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog. 2014, 10, e1004319. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gao, Y.; Zhang, G.; Li, D.; Wang, Z.; Zhang, J.; Hermida, L.C.; He, L.; Wang, Z.; Si, J.; et al. Enhancing KDM5A and TLR activity improves the response to immune checkpoint blockade. Sci. Transl. Med. 2020, 12, eaax2282. [Google Scholar] [CrossRef]
- Raung, S.L.; Chen, S.Y.; Liao, S.L.; Chen, J.H.; Chen, C.J. Japanese encephalitis virus infection stimulates Src tyrosine kinase in neuron/glia. Neurosci. Lett. 2007, 419, 263–268. [Google Scholar] [CrossRef]
- Józefiak, A.; Larska, M.; Pomorska-Mól, M.; Ruszkowski, J.J. The IGF-1 Signaling Pathway in Viral Infections. Viruses 2021, 13, 1488. [Google Scholar] [CrossRef]
- Das, S.; Chakraborty, S.; Basu, A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem. 2010, 115, 537–549. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Gupta, M.; Kumawat, K.L.; Basu, A. NLRP3 inflammasome: Key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE 2012, 7, e32270. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Ou, Y.C.; Chang, C.Y.; Pan, H.C.; Lin, S.Y.; Liao, S.L.; Raung, S.L.; Chen, S.Y.; Chang, C.J. Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem. Int. 2011, 58, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Das, S.; Joardar, S.; Bhattacharjee, S.; Chakraborty, P. Carotenoids as Anticancer Agents. In Carotenoids, Structure and Function in the Human Body; Springer: Cham, Switzerland, 2021; pp. 475–512. [Google Scholar]
- Muri, L.; Leppert, D.; Grandgirard, D.; Leib, S.L. MMPs and ADAMs in neurological infectious diseases and multiple sclerosis. Cell. Mol. Life. Sci. 2019, 76, 3097–3116. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Shakya, A.K.; Dhole, T.N.; Misra, U.K. Upregulated expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in BALB/c mouse brain challenged with Japanese encephalitis virus. Neuroimmunomodulation 2012, 19, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Shakya, A.K.; Shukla, M.; Kumari, N.; Krishnani, N.; Dhole, T.N.; Misra, U.K. Circulating levels of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases during Japanese encephalitis virus infection. Virusdisease 2016, 27, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Gerometta, R.; Kumar, S.; Shah, S.; Alvarez, L.; Candia, O.; Danias, J. Reduction of steroid-induced intraocular pressure elevation in sheep by tissue plasminogen activator. Invest. Ophthalmol. Vis. Sci. 2013, 54, 7903–7909. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, J.; Sun, H.; Xie, M.; Yang, C.; Pan, Y.; Huang, D.; Cheng, L.; Chen, H.; Ma, J.; et al. Autoimmune encephalitis after Japanese encephalitis in children: A prospective study. J. Neurol. Sci. 2021, 424, 117394. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F. Antibody-Mediated Encephalitis. N. Engl. J. Med. 2018, 378, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Ramberger, M.; O’Connor, K.C.; Bashford-Rogers, R.J.M.; Irani, S.R. The B cell immunobiology that underlies CNS autoantibody-mediated diseases. Nat. Rev. Neurol. 2020, 16, 481–492. [Google Scholar] [CrossRef]
- Tseng, Y.F.; Wang, C.C.; Liao, S.K.; Chuang, C.K.; Chen, W.J. Autoimmunity-related demyelination in infection by Japanese encephalitis virus. J. Biomed. Sci. 2011, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Getts, D.R.; Chastain, E.M.; Terry, R.L.; Miller, S.D. Virus infection; antiviral immunity; and autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.J.; Ou, Y.C.; Lin, S.Y.; Raung, S.L.; Liao, S.L.; Lai, C.Y.; Chen, S.Y.; Chen, J.H. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol. 2010, 91 Pt 4, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, R.; Ghildiyal, S.; Baluni, M.; Singh, D.; Srivastva, J.K.; Kumar, R.; Dhole, T.N. Association of interleukin-6 (174 G/C) and in-terleukin-12B (1188 A/C) gene polymorphism with expression and risk of Japanese encephalitis disease in North Indian population. J. Neuroimmunol. 2021, 358, 577630. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.M.; Dung, N.M.; Loan, H.T.; Kneen, R.; Wills, B.; le Thi, T.; House, D.; White, N.J.; Farrar, J.J.; Hart, C.A.; et al. Proinflam-matory cytokines and chemokines in humans with Japanese encephalitis. J. Infect. Dis. 2004, 190, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.J.; Ou, Y.C.; Li, J.R.; Chang, C.Y.; Pan, H.C.; Lai, C.Y.; Liao, S.L.; Raung, S.L.; Chang, C.J. Infection of pericytes in vitro by Japanese encephalitis virus disrupts the integrity of the endothelial barrier. J. Virol. 2014, 88, 1150–1161. [Google Scholar] [CrossRef] [Green Version]
- Brennan, K.; Zheng, J. Interleukin 8. In xPharm, The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; Volume 245, pp. 1–4. [Google Scholar]
- Singh, R.; Gautam, A.; Chandel, S.; Ghosh, A.; Dey, D.; Roy, S.; Ravichandiran, V.; Ghosh, D. Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2, An In Silico Study. Molecules 2020, 25, 4604. [Google Scholar] [CrossRef]
- Mehta, V.K.; Verma, R.; Garg, R.K.; Malhotra, H.S.; Sharma, P.K.; Jain, A. Study of interleukin-6 and interleukin-8 levels in patients with neurological manifestations of dengue. J. Postgrad. Med. 2017, 63, 11–15. [Google Scholar]
- Swarup, V.; Ghosh, J.; Duseja, R.; Ghosh, S.; Basu, A. Japanese encephalitis virus infection decrease endogenous IL-10 production, correlation with microglial activation and neuronal death. Neurosci. Lett. 2007, 420, 144–149. [Google Scholar] [CrossRef]
- Ghildiyal, S.; Fatima, T.; Singh, D.; Upadhyay, S.; Dhole, T.N.; Himanshu Reddy, D.; Kumar, A. Pro-inflammatory and an-ti-inflamatory cytokine genes polymorphisms and susceptibility to Japanese encephalitis disease in the North Indian population. Cytokine 2022, 149, 155716. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhen, Z.D.; Fan, D.Y.; Qin, C.F.; Han, D.S.; Zhou, H.N.; Wang, P.G.; An, J. Axl deficiency promotes the neuroinvasion of Japanese encephalitis virus by enhancing IL-1α production from pyroptotic macrophages. J. Virol. 2020, 94, e00602-20. [Google Scholar] [CrossRef]
- Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Adlakha, Y.K.; Basu, A. HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflammation 2018, 15, 1–9. [Google Scholar]
- Fan, S.; Yuan, J.; Deng, S.; Chen, Y.; Xie, B.; Wu, K.; Zhu, M.; Xu, H.; Huang, Y.; Yang, J.; et al. Activation of Interleukin-1β Release by the Classical Swine Fever Virus Is Dependent on the NLRP3 Inflammasome, Which Affects Virus Growth in Monocytes. Front. Cell. Infect. Microbiol. 2018, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.; Mathur, A.; Krishnani, N.; Dhole, T.N. An insufficient anti-inflammatory cytokine response in mouse brain is associated with increased tissue pathology and viral load during Japanese encephalitis virus infection. Arch. Virol. 2008, 153, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Tripathi, A.; Kumari, B.; Vrati, S.; Banerjee, A. Artificial MicroRNA-Mediated Inhibition of Japanese Encephalitis Virus Replication in Neuronal Cells. Nucleic Acid Ther. 2018, 28, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.; El-Kafrawy, S.A.; El-Day, M.M.; Alghafari, W.T.; Faizo, A.A.; Jha, S.K.; Dwivedi, V.D.; Azhar, E.I. Discovery of Small Molecules from Echinacea angustifolia Targeting RNA-Dependent RNA Polymerase of Japanese Encephalitis Virus. Life 2022, 12, 952. [Google Scholar] [CrossRef]
- Watson, A.A.; Lebedev, A.A.; Hall, B.A.; Fenton-May, A.E.; Vagin, A.A.; Dejnirattisai, W.; Felce, J.; Mongkolsapaya, J.; Palma, A.S.; Liu, Y.; et al. Structural flexibility of the macrophage dengue virus receptor CLEC5A, implications for ligand binding and signaling. J. Biol. Chem. 2011, 286, 24208–24218. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.J.; Antoshchenko, T.; Chen, Y.; Lu, H.; Pizarro, J.C.; Park, H.W. Probing the ATP Site of GRP78 with Nucleotide Triphos-phate Analogs. PLoS ONE 2016, 11, e0154862. [Google Scholar]
- Tojo, S.; Zhang, Z.; Matsui, H.; Tahara, M.; Ikeguchi, M.; Kochi, M.; Kamada, M.; Shigematsu, H.; Tsutsumi, A.; Adachi, N.; et al. Structural analysis reveals TLR7 dynamics underlying an-tagonism. Nat. Commun. 2020, 11, 5204. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef]
- Shakespeare, W.; Yang, M.; Bohacek, R.; Cerasoli, F.; Stebbins, K.; Sundaramoorthi, R.; Azimioara, M.; Vu, C.; Pradeepan, S.; Metcalf, C., 3rd; et al. Structure-based design of an osteoclast-selective; nonpeptide src homology 2 inhibitor with in vivo antiresorptive activity. Proc. Natl. Acad. Sci. USA 2000, 97, 9373–9378. [Google Scholar] [CrossRef] [Green Version]
- Porta, J.C.; Han, B.; Gulsevin, A.; Chung, J.M.; Peskova, Y.; Connolly, S.; Mchaourab, H.S.; Meiler, J.; Karakas, E.; Kenworthy, A.K.; et al. Molecular architecture of the human caveolin-1 complex. Sci. Adv. 2022, 8, eabn7232. [Google Scholar] [CrossRef]
- Poonsiri, T.; Wright, G.S.A.; Solomon, T.; Antonyuk, S.V. Crystal Structure of the Japanese Encephalitis Virus Capsid Protein. Viruses 2019, 11, 623. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, X.; Na, R.; Li, L.; Warkentin, E.; Witt, J.; Lu, X.; Yu, Y.; Wei, Y.; Peng, G.; et al. The structure differences of Japanese encephalitis virus SA14 and SA14-14-2 E proteins elucidate the virulence attenuation mechanism. Protein. Cell. 2019, 10, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Poonsiri, T.; Wright, G.S.A.; Diamond, M.S.; Turtle, L.; Solomon, T.; Antonyuk, S.V. Structural Study of the C-Terminal Domain of Nonstructural Protein 1 from Japanese Encephalitis Virus. J. Virol. 2018, 92, e01868-17. [Google Scholar] [CrossRef] [Green Version]
- Wahaab, A.; Liu, K.; Hameed, M.; Anwar, M.N.; Kang, L.; Li, C.; Ma, X.; Wajid, A.; Yang, Y.; Khan, U.H.; et al. Identification of Cleavage Sites Proteolytically Processed by NS2B-NS3 Protease in Polyprotein of Japanese Encephalitis Virus. Pathogens 2021, 10, 102. [Google Scholar] [CrossRef]
- Zheng, F.; Lu, G.; Li, L.; Gong, P.; Pan, Z. Uncoupling of Protease trans-cleavage and helicase activities in pestivirus NS3. J. Virol. 2017, 91, e01094-17. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, V.; Waltersperger, S.; Panepucci, E.; Chen, L.; Zhang, H.; Zhou, D.; Rose, J.; Ebihara, A.; Kuramitsu, S.; et al. Fast native-SAD phasing for routine macromolecular structure determination. Nat. Methods. 2015, 12, 131–133. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, M.; Zuo, Z. Overview of the anti-inflammatory effects; pharmacokinetic properties and clinical efficacies of arctigenin and arctiin from Arctium lappa L. Acta Pharmacol. Sin. 2018, 39, 787–801. [Google Scholar] [CrossRef]
- Luo, C.; Zou, L.; Sun, H.; Peng, J.; Gao, C.; Bao, L.; Ji, R.; Jin, Y.; Sun, S. A review of the anti-inflammatory effects of rosmarinic acid on inflammatory diseases. Front. Pharmacol. 2020, 11, 153. [Google Scholar] [CrossRef]
- Care, C.; Sornjai, W.; Jaratsittisin, J.; Hitakarun, A.; Wikan, N.; Triwitayakorn, K.; Smith, D.R. Discordant activity of kaempferol towards dengue virus and Japanese encephalitis virus. Molecules 2020, 25, 1246. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.G.; Li, Z.N.; Miao, X.W.; Li, S.; Zhu, H.L. Recent advances in natural products with antiviral activities. Mini. Rev. Med. Chem. 2021, 21, 1888–1908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lauwers, E.; Verstreken, P. Presynaptic protein homeostasis and neuronal function. Curr. Opin. Genet. Dev. 2017, 44, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Jia, X.; Liu, Y.; Wang, S.; Cao, J.; Zhang, B.; Xiao, G.; Wang, W. Screening of natural extracts for inhibitors against Japanese encephalitis virus infection. Antimicrob. Agents. Chemother. 2020, 64, e02373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.B.; Vrati, S.; Kalia, M. Pathobiology of Japanese encephalitis virus infection. Mol. Aspects. Med. 2021, 81, 100994. [Google Scholar] [CrossRef] [PubMed]
- Raung, S.L.; Chen, S.Y.; Liao, S.L.; Chen, J.H.; Chen, C.J. Tyrosine kinase inhibitors attenuate Japanese encephalitis virus-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2005, 327, 399–406. [Google Scholar] [CrossRef]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef]
- Fan, W.; Wu, M.; Qian, S.; Zhou, Y.; Chen, H.; Li, X.; Qian, P. TRIM52 inhibits Japanese Encephalitis Virus replication by degrading the viral NS2A. Sci. Rep. 2016, 6, 33698. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, J.F.; El-Gohary, Y.M. Nitazoxanide in the treatment of viral gastroenteritis, a randomized double-blind placebo-controlled clinical trial. Aliment Pharmacol. Ther. 2006, 24, 1423–1430. [Google Scholar] [CrossRef]
- Patil, G.; Li, S. Tripartite motif proteins, an emerging antiviral protein family. Future Virol. 2019, 14, 107–122. [Google Scholar] [CrossRef]
- Dwivedi, V.D.; Singh, A.; El-Kafraway, S.A.; Alandijany, T.A.; Faizo, A.A.; Bajrai, L.H.; Kamal, M.A.; Azhar, E.I. Mechanistic insights into the Japanese encephalitis virus RNA dependent RNA polymerase protein inhibition by bioflavonoids from Azadirachta in-dica. Sci. Rep. 2021, 11, 18125. [Google Scholar] [CrossRef]
- Fang, J.; Sun, L.; Peng, G.; Xu, J.; Zhou, R.; Cao, S.; Chen, H.; Song, Y. Identification of three antiviral inhibitors against Japanese en-cephalitis virus from library of pharmacologically active compounds 1280. PLoS ONE 2013, 8, e78425. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, L.; Madhusudana, S.N.; Ravi, V.; Desai, A. Mycophenolic acid inhibits replication of Japanese encephalitis virus. Chemotherapy 2011, 57, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.H.; Huang, Y.L.; Chen, C.C.; Pi, W.C.; Hsu, Y.L.; Lo, L.C.; Chen, W.Y.; Fu, S.L.; Lin, C.H. Andrographolide and its fluorescent derivative inhibit the main proteases of 2019-nCoV and SARS-CoV through covalent linkage. Biochem. Biophys. Res. Commun. 2020, 533, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Konishi, E. Potential chemotherapeutic targets for Japanese encephalitis, current status of antiviral drug development and future challenges. Expert Opin. Ther. Targets. 2015, 19, 1379–1395. [Google Scholar] [CrossRef]
- Lin, C.W.; Wu, C.F.; Hsiao, N.W.; Chang, C.Y.; Li, S.W.; Wan, L.; Lin, Y.J.; Lin, W.Y. Aloe-emodin is an interferon-inducing agent with antiviral activity against Japanese encephalitis virus and enterovirus 71. Int. J. Antimicrob. Agents 2008, 32, 355–359. [Google Scholar] [CrossRef]
- Kumar, R.; Tripathi, P.; Baranwal, M.; Singh, S.; Tripathi, S.; Banerjee, G. Randomized; controlled trial of oral ribavirin for Japanese encephalitis in children in Uttar Pradesh; India. Clin. Infect. Dis. 2009, 48, 400–406. [Google Scholar] [CrossRef]
- Nagarakanti, S.; Bishburg, E. Is minocycline an antiviral agent? A review of current literature. Basic Clin. Pharmacol. Toxicol. 2016, 118, 4–8. [Google Scholar] [CrossRef]
- Dutta, K.; Ghosh, D.; Basu, A. Curcumin protects neuronal cells from Japanese encephalitis virus-mediated cell death and also inhibits infective viral particle formation by dysregulation of ubiquitin-proteasome system. J. Neuroimmune Pharmacol. 2009, 4, 328–337. [Google Scholar] [CrossRef]
- Fan, W.; Qian, S.; Qian, P.; Li, X. Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016, 220, 112–116. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Y.; Yan, Y.; Li, W.; Zhao, L.; Dai, Q.; Li, Y.; Li, S.; Zhong, J.; Cao, R.; et al. Small molecule inhibitor of atpase activity of hsp70 as a broad-spectrum inhibitor against flavivirus infections. ACS Infect. Dis. 2020, 6, 832–843. [Google Scholar] [CrossRef]
- Nunes, D.A.F.; Santos, F.R.D.S.; da Fonseca, S.T.D.; de Lima, W.G.; Nizer, W.S.D.C.; Ferreira, J.M.S.; de Magalhães, J.C. NS2B-NS3 protease inhibitors as promising compounds in the development of antivirals against Zika virus, A systematic review. J. Med. Virol. 2022, 94, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Ou, Y.C.; Raung, S.L.; Chen, C.J. Antiviral effect of dehydroepiandrosterone on Japanese encephalitis virus infection. J. Gen. Virol. 2005, 86 Pt 9, 2513–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Liu, K.; Miao, D.; Cao, R.; Chen, P. Effective inhibition of Japanese encephalitis virus replication by shRNAs targeting various viral genes in vitro and in vivo. Virology 2014, 454, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Zu, X.; Liu, Y.; Wang, S.; Jin, R.; Zhou, Z.; Liu, H.; Gong, R.; Xiao, G.; Wang, W. Peptide inhibitor of Japanese encephalitis virus infection targeting envelope protein domain III. Antivir. Res. 2014, 104, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Maurya, V.K.; Kabir, R.; Nayak, D.; Khurana, A.; Manchanda, R.K.; Gadugu, S.; Shanker, K.; Saxena, S.K. Antiviral activity of belladonna during Japanese encephalitis virus infection via inhibition of microglia activation and inflammation leading to neuronal cell survival. ACS Chem. Neurosci. 2020, 11, 3683–3696. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.J.; Park, S.N.; Oh, J.W. Antiviral effect of amphotericin B on Japanese encephalitis virus replication. J. Microbiol. Biotechnol. 2004, 14, 121–127. [Google Scholar]
- Joe, S.; Salam, A.A.A.; Neogi, U.; Babu, N.N.; Mudgal, P.P. Antiviral drug research for Japanese encephalitis, an updated review. Pharmacol. Rep. 2022, 74, 273–296. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| S. No. | Groups | Rate of Infection and Vaccination Programmes | Countries or Regions | Incidences/100,000 | Case Frequency Ratio (Child: Adult) | References |
|---|---|---|---|---|---|---|
| 1 | A | Vaccination programs of high quality in high-incidence areas | Japan, the Republic of Korea, China Taiwan | 0.003 | 07:01 | [20,21,22] |
| 2 | B | Areas with extremely low incidence and no immunization programs | Australia, Pakistan, Russia, Singapore | 0.003 | 07:01 | [23,24] |
| 3 | C | Areas with extremely low incidence and no immunization programs | China | 3.3 | 03:01 | [25,26] |
| 4 | D | Vaccination programs are weak or non-existent in high-incidence regions. | Cambodia, Indonesia, Laos, Malaysia, Myanmar, Philippines, Timor-Leste | 10.6 | 07:01 | [27,28] |
| 5 | E | Areas with a medium incidence but no immunization programs | Malaysia, Papua New Guinea | 5.3 | NA | [29] |
| 6 | F | Vaccination programs are being expanded in high-incidence regions. | India, Nepal | 2.8 | 05:04 | [22,30] |
| 7 | G | Vaccination programs are inadequate or non-existent in low-incidence areas. | Bangladesh, Bhutan, Brunei, Nepal (lower incidence stratum) | 1 | 04:01 | [31,32] |
| 8 | H | Areas with a medium to a high frequency of disease and growing immunization programs | India (medium incidence stratum), Malaysia (Sarawak), the Republic of Korea, Sri Lanka, Thailand, Vietnam | 1.5 | 07:01 | [24,33,34] |
| Countries or Regions | First Reported | Outbreaks | Diagnosis | Treatments | JE Incidence Rate | Programmes Run by the Governments | References |
|---|---|---|---|---|---|---|---|
| Australia | 1995 | 2 in 1995, 2 in 1998, and 1 in 2022 | Viral antigen detection, JEV-specific antibody detection, reverse passive hemagglutination, staphylococcal co-agglutination tests, ELISA, qPCR, RT-PCR, RT-LAMP | MBDV, Fever relief medicines, plenty of fluid | 3 cases | 69 million dollar program to control JEV infection, including mosquito control, vaccination and sentinel pig surveillance programs | [35] |
| Bangladesh | 1977 | 22 patients with 7 deaths in 1977 | Viral antigen detection, JEV-specific antibody detection, ELISA, qPCR, RT-PCR | Fever and pain relief medicines, plenty of fluid | 0.6–2.7 cases/lakh | - | [9,36] |
| Bhutan | - | - | ELISA, PCR | Fever and pain relief medicines, fluid | - | Integrated national vaccination program | [8] |
| Brunei | - | - | ELISA, PCR | Fever and pain relief medicine, plenty of fluid | - | - | [13] |
| Cambodia | 1947 | - | ELISA, PCR | LAV-SA14-14-2 vaccine, fever and pain relief medicines, plenty of fluid | 11.1 cases/lakh | National vaccination program | [13] |
| China | 1940s | 1960–1970 with morbidity >10 cases/lakh | Viral antigen detection, JEV-specific antibody detection, reverse passive hemagglutination, staphylococcal co-agglutination tests, ELISA, qPCR, RT-PCR, RT-LAMP | LAV-SA14-14-2 vaccine, MBDV, fever and pain relief medicines, plenty of fluid | 0.1–0.9 cases/lakh | Expanded program on immunization to reduce JE | [37] |
| Guam | 1947 | 46 reported cases | - | Fever relief medicines, plenty of fluid | - | - | [13] |
| India | 1950 | 5700 cases with 1315 deaths in 2005 | Viral antigen detection, JEV-specific antibody detection, reverse passive hemagglutination, staphylococcal co-agglutination tests, ELISA, qPCR, RT-PCR, RT-LAMP | LAV-SA14-14-2 vaccine, fever and pain relief medicines, plenty of fluid | 15 cases/lakh | Government immunization program | [38] |
| Indonesia | 1974 | - | JEV-specific antibody detection, ELISA, PCR, RT-PCR | Fever and pain relief medicines, plenty of fluid | 8.2 cases/lakh | No vaccination program | [9,39] |
| Japan | 1933 | Mainly before 1960 | Viral antigen detection, JEV-specific antibody detection, reverse passive hemagglutination, staphylococcal co-agglutination tests, ELISA, qPCR, RT-PCR | VCDV-Bejing-I vaccine, pain and fever relief medicines | <10 cases | Vaccination program | [9] |
| Laos | 1989 | - | JEV-specific antibody detection in the CSF | Fever and pain relief medicines, fluids | - | JEV vaccination in 2013 | [40] |
| Malaysia | 1952 | 154 cases with 42 deaths in 1999 | Viral antigen detection, JEV-specific antibody detection, ELISA, qPCR | MBDV, fever relief medicines | 4.3 cases/lakh | JE vaccination was introduced in July 2001, and the vaccination was only practiced in Sarawak and formalin-activated mouse-derived JE vaccine (Biken, Japan) is used in Malaysia | [41] |
| Myanmar | 1968 | 5 cases with 4 deaths in 1947 and 43 with 32 deaths in 1948 | Viral antigen detection, JEV-specific antibody detection, ELISA, qPCR, RT-PCR | Fever and pain relief medicines, plenty of fluid | - | National vaccination program | [9,13] |
| Nepal | 1978 | 2040 cases with 205 deaths in 2005 | JEV-specific antibody detection, ELISA | LASV-SA14-14-2 vaccine, fever relief medicines | 1.3 cases/lakh | National JE prevention and control program in Kathmandu valley | [31] |
| Pakistan | 1980s | - | JEV-specific antibody detection | Fever relief medicines | - | No vaccination programs | [9] |
| Papua New Guinea | 1995 | - | JEV-specific antibody detection, ELISA, PCR | Fever relief medicines | - | - | [42] |
| Philippines | 1950s | - | - | - | - | - | [13] |
| Saipan, USA | 1990 | 1990 | JEV-specific antibody detection in CSF, ELISA | Fever relief medicines | - | no vaccination programs | [43] |
| Singapore | 1952 | - | Viral antigen detection, JEV-specific antibody detection, reverse passive hemagglutination, staphylococcal co-agglutination tests, ELISA, qPCR, RT-PCR | Fever relief medicines | <5 cases | Vaccination programs reduce JE case | [9,13] |
| Republic of Korea | 1946 | - | JEV-specific antibody detection in serum and CSF, ELISA, PRNT, PCR | MBDV, fever relief medicines | 0.01–0.08 cases/lakh and <10 cases/year | Successful vaccination program for three decades in the Republic of Korea | [44] |
| Sri Lanka | 1968 | - | ELISA, PCR | Fever relief medicines | <100 cases | JE vaccination program | [9,13] |
| China Taiwan | 1938 | 1960–1970 with morbidity ~12.4 cases/lakh | Viral antigen detection, JEV-specific antibody detection, ELISA, qPCR, RT-PCR | MBDV, Fever relief medicines, plenty of fluid | 0.03/lakh | Taiwan National Infectious Disease Statistics System–Japanese Encephalitis, Self-reporting through the toll-free 1922 hotline or local public health authority. | [45] |
| Thailand | 1961 | - | RT-PCR | MBD vaccine, fever relief medicines | 300 cases/year | vaccination program | [9,13] |
| Vietnam | 1960 | - | - | MBDV, fever relief medicines | 1–8 cases/lakh | vaccination program (from 1997) | [9,13] |
| S. No. | Name of Targets | PDB ID | Ligands | Amino Acid Residues of Active Sites | Resolution of Crystal Structures | References |
|---|---|---|---|---|---|---|
| 1 | CLEC5A | 2YHF | NA | - | 1.9 Å | [130] |
| 2 | GRP78 | 5F1X | ATP | Thr37, Thr38, Tyr39, Gly227, Gly228, Thr229, Glu293, Lys296, Ser300, Gly364, | 1.9 Å | [131] |
| 3 | TLR7 | 6LW1 | RIJUCCOLHSAZPO-GOTSBHOMSA-N | Asn265, Tyr264, Phe349, Glu352, Leu353, Gln354, Val355, Val381, Thr406, Phe408, Phe507, Ser530, Gln531 | 2.8 Å | [132] |
| 4 | D2R | 7DFP | DKGZKTPJOSAWFA-UHFFFAOYSA-N | Val91, Leu94, Val111, Asp114, Val115, Cys118, Cys182, Ile184, Trp386, Phe390, Thr412, Tyr416, | 3.1 Å | [133] |
| 5 | Src | 1FBZ | SPSGYTWOIGAABK-DQEYMECFSA-N | Arg12, Arg32, Glu35, Ser36, His58, Lys60 | 2.4 Å | [134] |
| 6 | Caveolin-1 | 7SC0 | NA | - | 3.4 Å | [135] |
| 7 | Capsid | 5OW2 | KRKNYBCHXYNGOX-UHFFFAOYSA-K | Pro43, Val44, | 1.98 Å | [136] |
| 8 | Envelop | 5MV1 | NA | - | 2.25 Å | [137] |
| 9 | NS1 | 5O36 | QAOWNCQODCNURD-UHFFFAOYSA-L | Arg347, Gln349 | 2.6 Å | [138] |
| 10 | NS3 | 2Z83 | NA | - | - | [139] |
| 11 | NS5(RdRp) | 4HDH | ZKHQWZAMYRWXGA-KQYNXXCUSA-N | Arg460, Arg474, Asp668, Ser715, Arg734, Arg742, Ser799, Trp800, | 2.28 Å | [140] |
| 12 | NS3/NS4A | 5WX1 | NA | - | 2.35 Å | [140] |
| 13 | NS2b-NS3 | 4R8T | VEXZGXHMUGYJMC-UHFFFAOYSA-M | Gly151 | 2.133 Å | [141] |
| S. No. | Nature-Derived Compounds | Mechanisms | References |
|---|---|---|---|
| 1 | Arctigenin | Decreases JEV-induced neuronal apoptosis, microglial activation, and caspase activity. | [142] |
| 2 | Baicalein | Extracellular virucidal activity. | [145] |
| 3 | Cilnidipine | Inhibits JEV in high-throughput screening assay (HTS) with EC50 of 6.52 µM. | [155] |
| 4 | Cinaroside | Inhibits non-structural protein (RdRp) in silico. | [129] |
| 5 | Digoxin | Reported to act as an inhibitor of the Na+/K+-ATPase pump. | [146] |
| 6 | Echinacin | Inhibits RdRp in silico. | [129] |
| 7 | Echinacoside | Inhibits RdRp in silico. | [129] |
| 8 | FGIM-1-27 | Inhibits JEV in high-throughput screening assay (HTS) with EC50 s of 3.21 µM. | [155] |
| 9 | Gedunin | Inhibits RdRp in silico. | [154] |
| 10 | Genistein | Reduces the effect of neurotoxicity induced by JEV and suppresses the cachectin (TNF-α) and leukocytic pyrogen (IL-1β) prompted by JEV at the transcriptional level. | [149] |
| 11 | Herbimycin A | Reduces the effect of neurotoxicity induced by JEV and suppresses the cachectin (TNF-α) and leukocytic pyrogen (IL-1β) prompted by JEV at the transcriptional level. | [149] |
| 12 | IFIT 1 | Inhibits JEV replication by binding to the 5′ -triphosphate RNA and, most preferably, to the 5′ capped 2′-O unmethylated mRNA. | [150] |
| 13 | Kaempferol-3-glucoside | Inhibits RdRp in silico. | [129] |
| 14 | Kulactone | Inhibits RdRp in silico. | [154] |
| 15 | Manidipine | Inhibits intracellular Ca2+, which is required for JEV entry, replication, and budding. | [149] |
| 16 | Mycophenolic acid | Reported antiviral activity of an immune suppressant as an anti-JEV drug via plaque reduction neutralization assay, virus yield reduction assay, and cytopathic effect inhibition assay, accompanied by an IC50 of 3.1 µg/mL through in vivo and in vitro experiments. | [156] |
| 17 | Niclosamide | Inhibits JEV with EC50 of 5.80 µM | [155] |
| 18 | Nimbolide | Inhibits RdRp in silico. | [154] |
| 19 | Nitazoxanide | Inhibits the replication machinery, validated through both in vivo as well as in vitro methods, which suggests this compound is a potential agent for JE treatment. | [152,157] |
| 20 | Ohchinin acetate | Inhibits non-structural protein (RdRp) in silico. | [154] |
| 21 | Ouabain | Reported against the Na+/K+-ATPase as an inhibitor during the replication of the JEV in the BALB/C mouse model | [147] |
| 22 | PP2 | Reduces the effect of neurotoxicity induced by JEV and suppresses the Cachectin (TNF-α) and leukocytic pyrogen (IL-1β) prompted by JEV at the transcriptional level. | [149] |
| 23 | Quercetagetin 7-glucoside | Inhibits RdRp in silico. | [129] |
| 24 | Rosmarinic acid | Reduces induction of proinflammatory mediators, neuronal apoptosis, microglial activation, and caspase activation. | [143] |
| 25 | Rutin | Inhibits RdRp in silico. | [129] |
| 26 | TRIM52 | NS2A was degraded by TRIM 52 within a proteosome-dependent process through E3 Ubiquitin synthetase activity. Overexpression of TRIM52 in BHK-21 cells directly shows E3 Ubiquitin ligase activity and activation of the host innate immune system. | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, P.; Chakraborty, P.; Jha, N.K.; Dewanjee, S.; Jha, A.K.; Panda, S.P.; Mishra, P.C.; Dey, A.; Jha, S.K. Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases. Viruses 2022, 14, 2686. https://doi.org/10.3390/v14122686
Yadav P, Chakraborty P, Jha NK, Dewanjee S, Jha AK, Panda SP, Mishra PC, Dey A, Jha SK. Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases. Viruses. 2022; 14(12):2686. https://doi.org/10.3390/v14122686
Chicago/Turabian StyleYadav, Pardeep, Pratik Chakraborty, Niraj Kumar Jha, Saikat Dewanjee, Abhimanyu Kumar Jha, Siva Prasad Panda, Prabhu Chandra Mishra, Abhijit Dey, and Saurabh Kumar Jha. 2022. "Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases" Viruses 14, no. 12: 2686. https://doi.org/10.3390/v14122686
APA StyleYadav, P., Chakraborty, P., Jha, N. K., Dewanjee, S., Jha, A. K., Panda, S. P., Mishra, P. C., Dey, A., & Jha, S. K. (2022). Molecular Mechanism and Role of Japanese Encephalitis Virus Infection in Central Nervous System-Mediated Diseases. Viruses, 14(12), 2686. https://doi.org/10.3390/v14122686