
ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection

 , , and
, , and 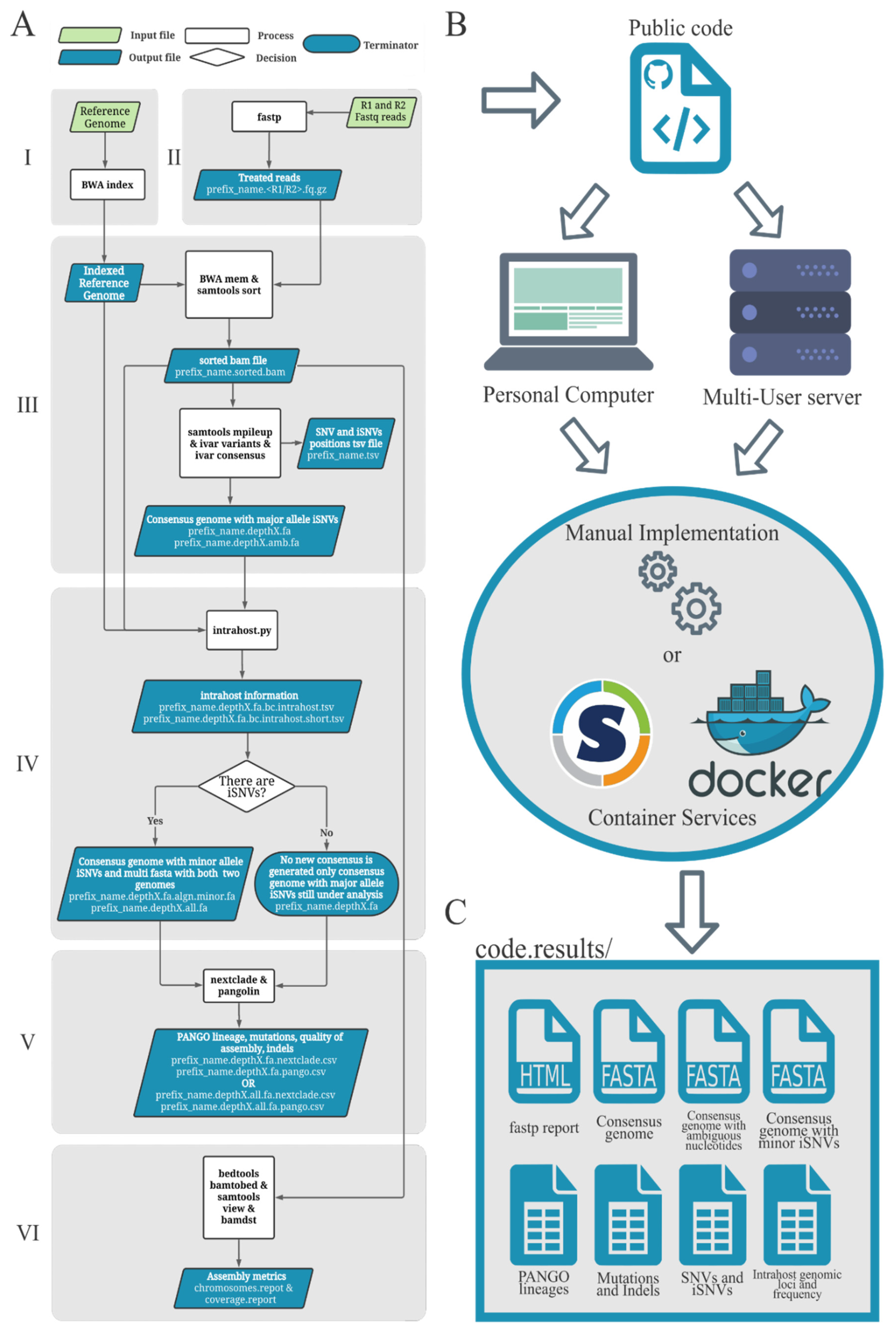{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Worflow Structure
- (i)
- The minor allele frequency represented at least 5% of the total allele depth;
- (ii)
- The minor alleles had at least 100 reads of depth (default depth);
- (iii)
- The minor allele nucleotides were supported by reads of both senses (at least 5% of depth should come from each read sense).
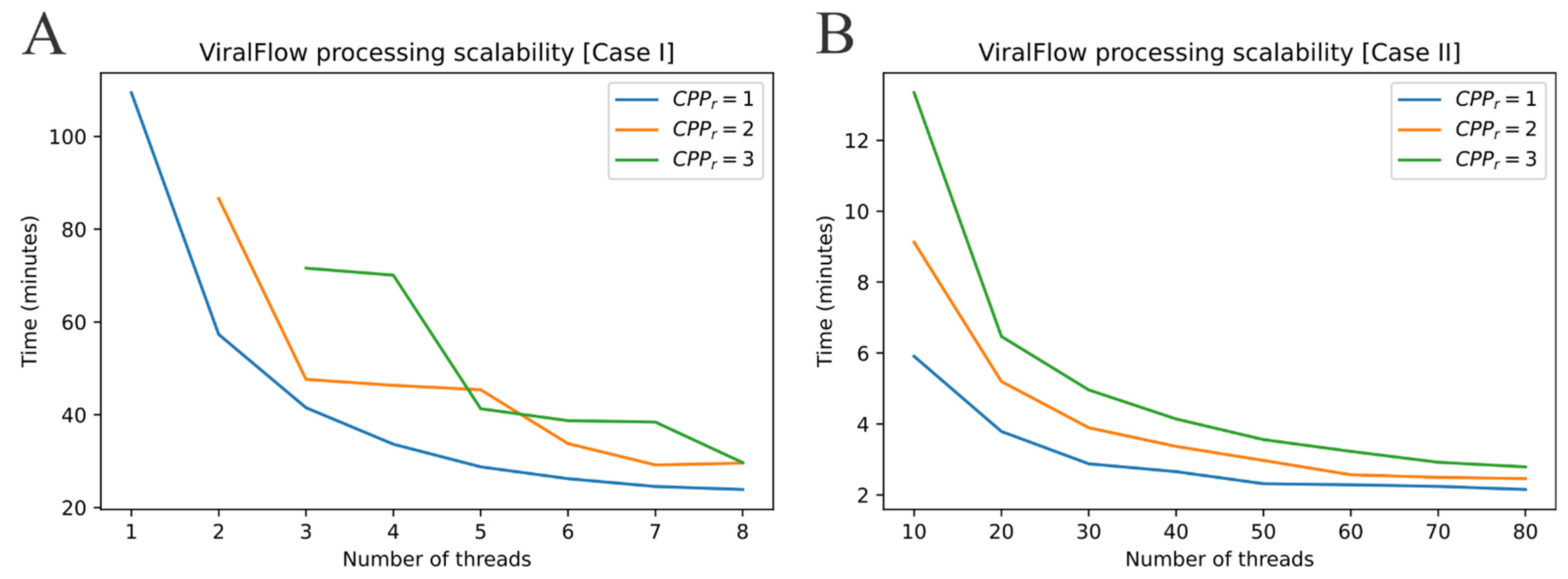
2.2. ViralFlow Scalability
- Case I: Using an average personal computer to install all dependencies or using Docker or Singularity container services.
- Case II: Using a multi-user computational server to install all dependencies or using Docker or Singularity container services.
2.3. Benchmark
3. Results and Discussion
3.1. Performance and Scalability
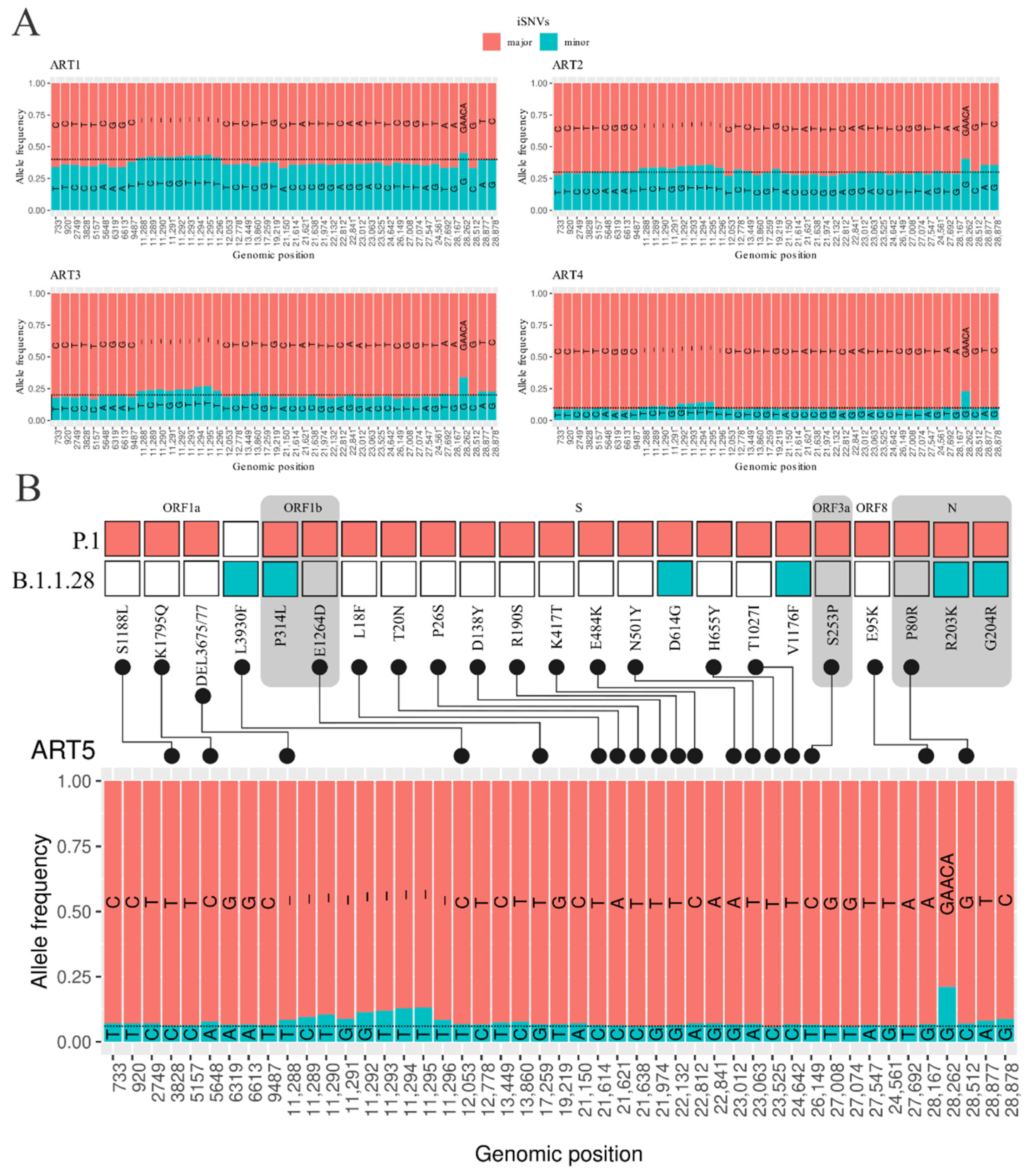
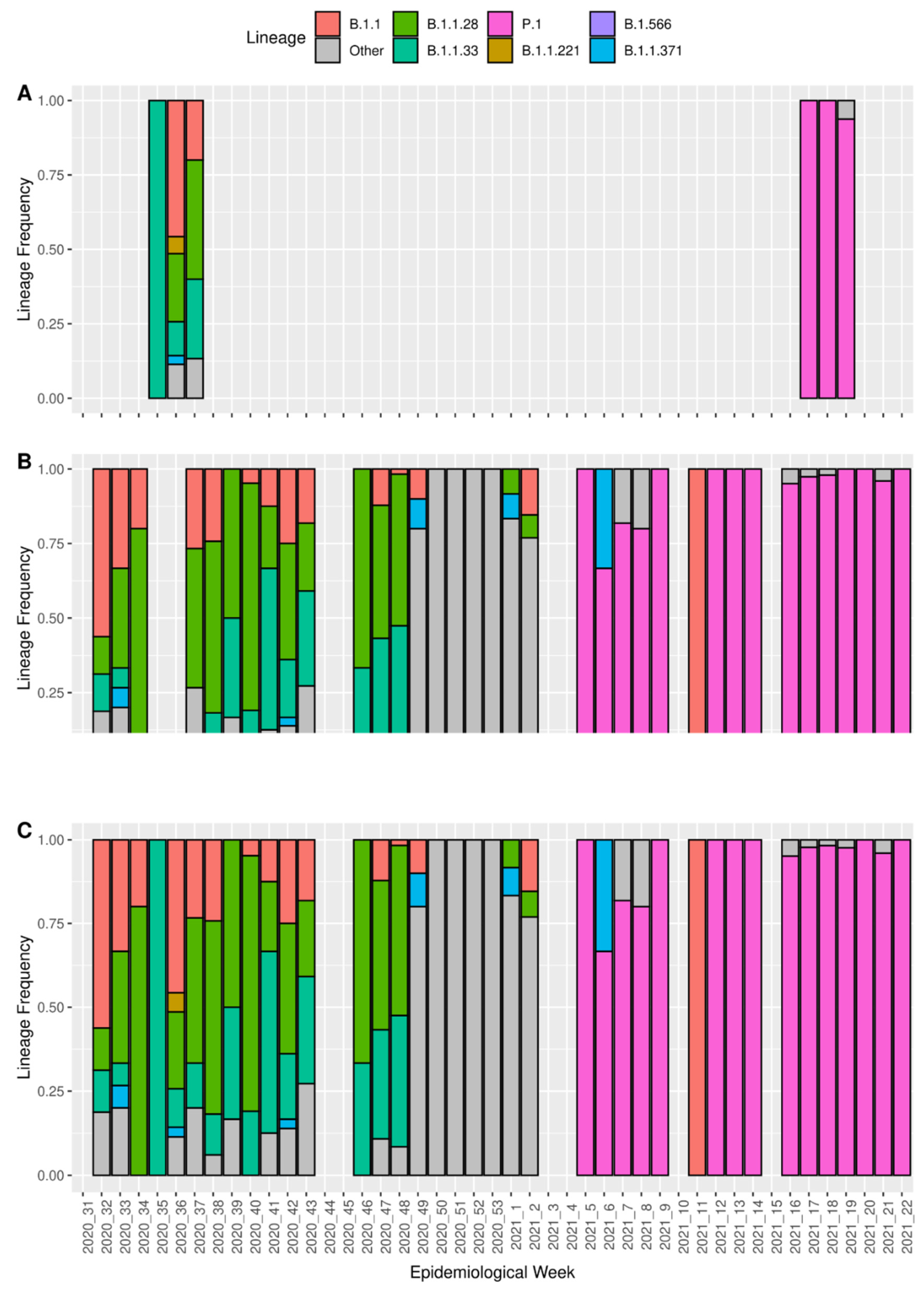
3.2. Intrahost Detection
3.3. Detection of Coinfection Events
3.4. Additional Quality-Check Results
3.5. Benchmark
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 27 September 2021).
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’ Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCRone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.J.R.; Silva, C.T.A.d.; Guarines, K.M.; Mendes, R.P.G.; Pardee, K.; Kohl, A.; Pena, L. Clinical and Laboratory Diagnosis of SARS-CoV-2, the Virus Causing COVID-19. ACS Infect. Dis. 2020, 6, 2319–2336. [Google Scholar] [CrossRef] [PubMed]
- Nörz, D.; Grunwald, M.; Olearo, F.; Fischer, N.; Aepfelbacher, M.; Pfefferle, S.; Lütgehetmann, M. Evaluation of a Fully Automated High-Throughput SARS-CoV-2 Multiplex QPCR Assay with Built-in Screening Functionality for Del-HV69/70- and N501Y Variants Such as B.1.1.7. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2021, 141, 104894. [Google Scholar] [CrossRef]
- Bezerra, M.F.; Machado, L.C.; De Carvalho, V.d.C.V.; Docena, C.; Brandão-Filho, S.P.; Ayres, C.F.J.; Paiva, M.H.S.; Wallau, G.L. A Sanger-Based Approach for Scaling up Screening of SARS-CoV-2 Variants of Interest and Concern. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 92, 104910. [Google Scholar] [CrossRef]
- Alves, P.A.; de Oliveira, E.G.; Franco-Luiz, A.P.M.; Almeida, L.T.; Gonçalves, A.B.; Borges, I.A.; de Souza Rocha, F.; Rocha, R.P.; Bezerra, M.F.; Miranda, P.; et al. Optimization and Clinical Validation of Colorimetric Reverse Transcription Loop-Mediated Isothermal Amplification, a Fast, Highly Sensitive and Specific COVID-19 Molecular Diagnostic Tool That Is Robust to Detect SARS-CoV-2 Variants of Concern. Front. Microbiol. 2021, 12, 713713. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-Based Approaches for SARS-CoV-2 Whole Genome Characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef]
- Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Mattison, S.; Gazy, I.; Fish, M.; et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes 2020, 11, 949. [Google Scholar] [CrossRef] [PubMed]
- Nanopore Sequencing the SARS-CoV-2 Genome: Introduction to Protocol. Available online: http://nanoporetech.com/resource-centre/nanopore-sequencing-sars-cov-2-genome-introduction-protocol (accessed on 25 November 2021).
- Campos, G.S.; Sardi, S.I.; Falcao, M.B.; Belitardo, E.M.M.A.; Rocha, D.J.P.G.; Rolo, C.A.; Menezes, A.D.; Pinheiro, C.S.; Carvalho, R.H.; Almeida, J.P.P.; et al. Ion Torrent-Based Nasopharyngeal Swab Metatranscriptomics in COVID-19. J. Virol. Methods 2020, 282, 113888. [Google Scholar] [CrossRef] [PubMed]
- SARS-CoV-2 Sequencing Resources; Centers for Disease Control and Prevention. 2021. Available online: https://github.com/CDCgov/SARS-CoV-2_Sequencing (accessed on 27 September 2021).
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Hölzer, M. PoreCov-An Easy to Use, Fast, and Robust Workflow for SARS-CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2021, 12, 711437. [Google Scholar] [CrossRef]
- ARTIC. ARTICnetwork. 2021. Available online: https://github.com/artic-network/fieldbioinformatics (accessed on 29 December 2021).
- RKIBioinformaticsPipelines/CoVpipe. Available online: https://gitlab.com/RKIBioinformaticsPipelines/ncov_minipipe (accessed on 29 December 2021).
- Resende, P.C.; Naveca, F.G.; Lins, R.D.; Dezordi, F.Z.; Ferraz, M.V.F.; Moreira, E.G.; Coêlho, D.F.; Motta, F.C.; Paixão, A.C.D.; Appolinario, L.; et al. The Ongoing Evolution of Variants of Concern and Interest of SARS-CoV-2 in Brazil Revealed by Convergent Indels in the Amino (N)-Terminal Domain of the Spike Protein. Virus Evol 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Paiva, M.H.S.; Guedes, D.R.D.; Docena, C.; Bezerra, M.F.; Dezordi, F.Z.; Machado, L.C.; Krokovsky, L.; Helvecio, E.; da Silva, A.F.; Vasconcelos, L.R.S.; et al. Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses 2020, 12, 1414. [Google Scholar] [CrossRef] [PubMed]
- Empowering App Development for Developers|Docker. Available online: https://www.docker.com/ (accessed on 27 September 2021).
- Singularity. Available online: https://sylabs.io/singularity/ (accessed on 27 September 2021).
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinform. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinform. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Khanna, A.; Larson, D.E.; Srivatsan, S.N.; Mosior, M.; Abbott, T.E.; Kiwala, S.; Ley, T.J.; Duncavage, E.J.; Walter, M.J.; Walker, J.R.; et al. Bam-Readcount—Rapid Generation of Basepair-Resolution Sequence Metrics. arXiv 2021, arXiv:2107.12817v1. [Google Scholar]
- Aksamentov, I.; Neher, R. Nextclade. Available online: https://clades.nextstrain.org (accessed on 2 September 2021).
- Quan, S. Bamdst—A BAM Depth Stat. Tool. 2021. Available online: https://github.com/shiquan (accessed on 27 September 2021).
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong Nguyen, P.T.; Plyusnin, I.; Sironen, T.; Vapalahti, O.; Kant, R.; Smura, T. HAVoC, a Bioinformatic Pipeline for Reference-Based Consensus Assembly and Lineage Assignment for SARS-CoV-2 Sequences. BMC Bioinform. 2021, 22, 373. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome–Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosterhout, C.; Hall, N.; Ly, H.; Tyler, K.M. COVID-19 Evolution during the Pandemic—Implications of New SARS-CoV-2 Variants on Disease Control and Public Health Policies. Virulence 2021, 12, 507–508. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; et al. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dezordi, F.Z.; Neto, A.M.d.S.; Campos, T.d.L.; Jeronimo, P.M.C.; Aksenen, C.F.; Almeida, S.P.; Wallau, G.L.; on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses 2022, 14, 217. https://doi.org/10.3390/v14020217
Dezordi FZ, Neto AMdS, Campos TdL, Jeronimo PMC, Aksenen CF, Almeida SP, Wallau GL, on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses. 2022; 14(2):217. https://doi.org/10.3390/v14020217
Chicago/Turabian StyleDezordi, Filipe Zimmer, Antonio Marinho da Silva Neto, Túlio de Lima Campos, Pedro Miguel Carneiro Jeronimo, Cleber Furtado Aksenen, Suzana Porto Almeida, Gabriel Luz Wallau, and on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. 2022. "ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection" Viruses 14, no. 2: 217. https://doi.org/10.3390/v14020217
APA StyleDezordi, F. Z., Neto, A. M. d. S., Campos, T. d. L., Jeronimo, P. M. C., Aksenen, C. F., Almeida, S. P., Wallau, G. L., & on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. (2022). ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses, 14(2), 217. https://doi.org/10.3390/v14020217