
The Synthesis and Anti-Cytomegalovirus Activity of Piperidine-4-Carboxamides




Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Viruses
2.3. Cell Culture, Virus Infection, and Anti-Viral Assays
2.4. Toxicity Assays
2.5. SDS-PAGE and Immunoblot Analysis
2.6. DNA Isolation and Quantitative Real-Time (qPCR)
2.7. Add-On and Removal Assays
2.8. Combination Assays
2.9. Statistical Analysis
3. Results
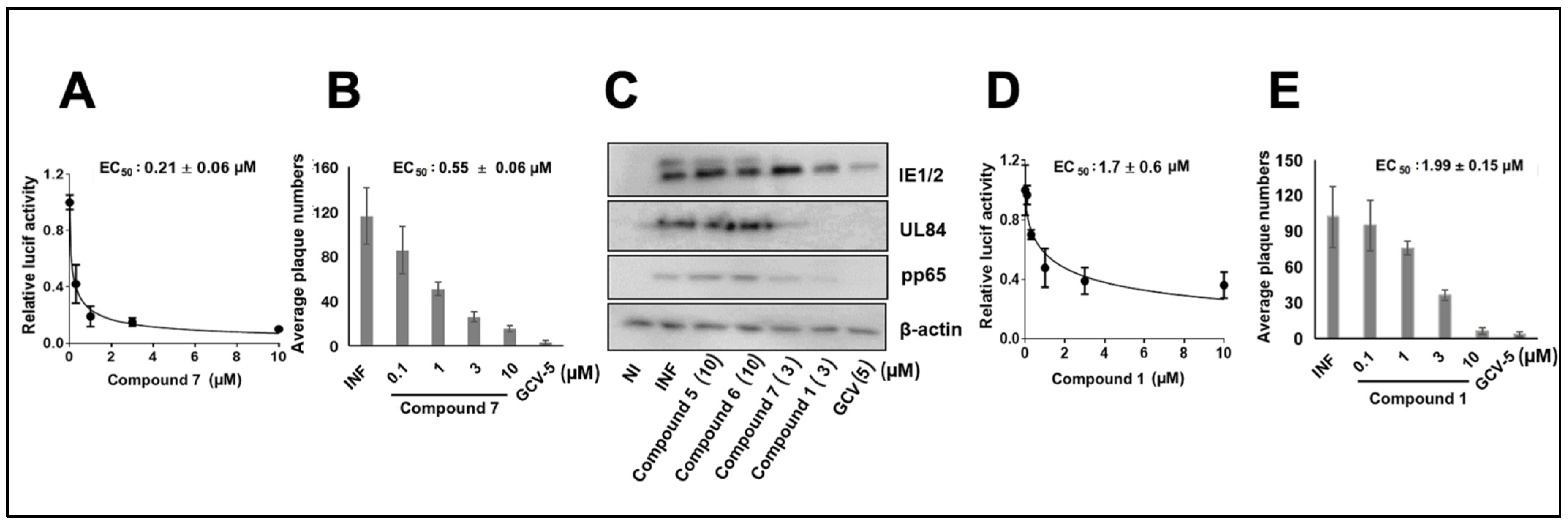
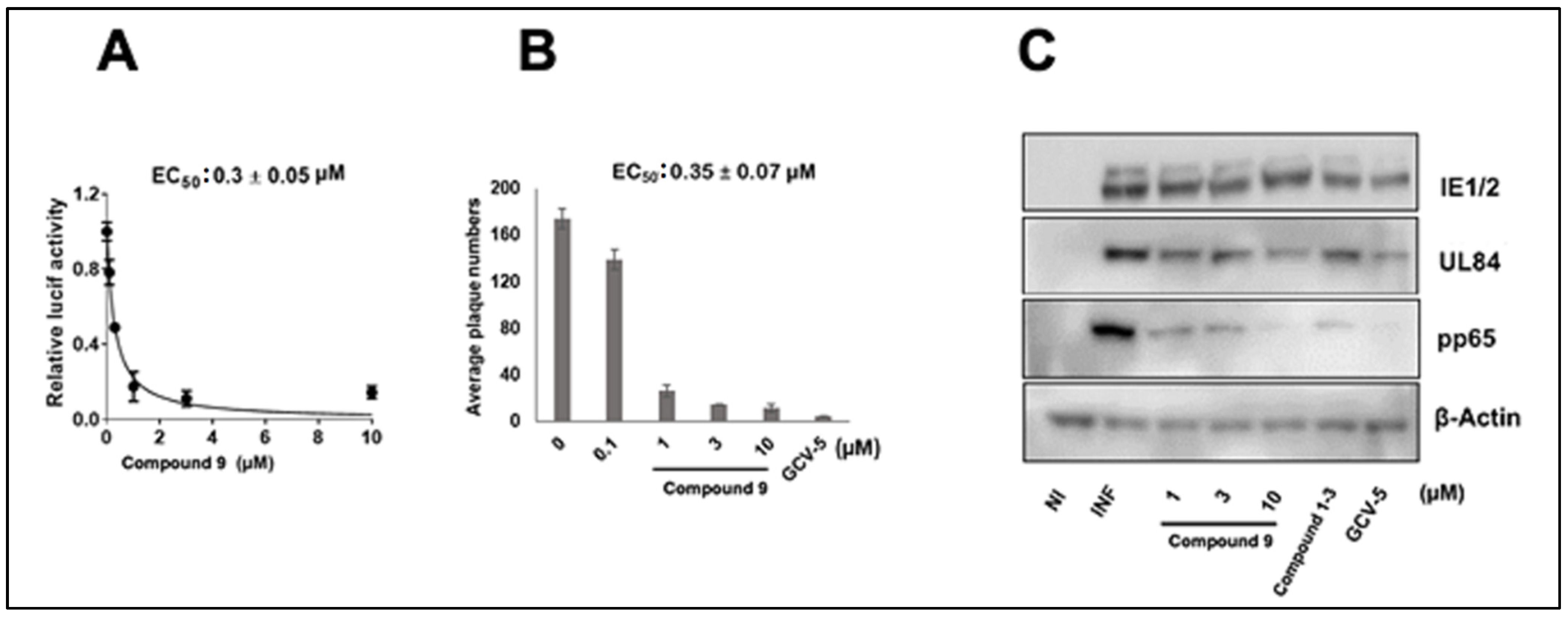
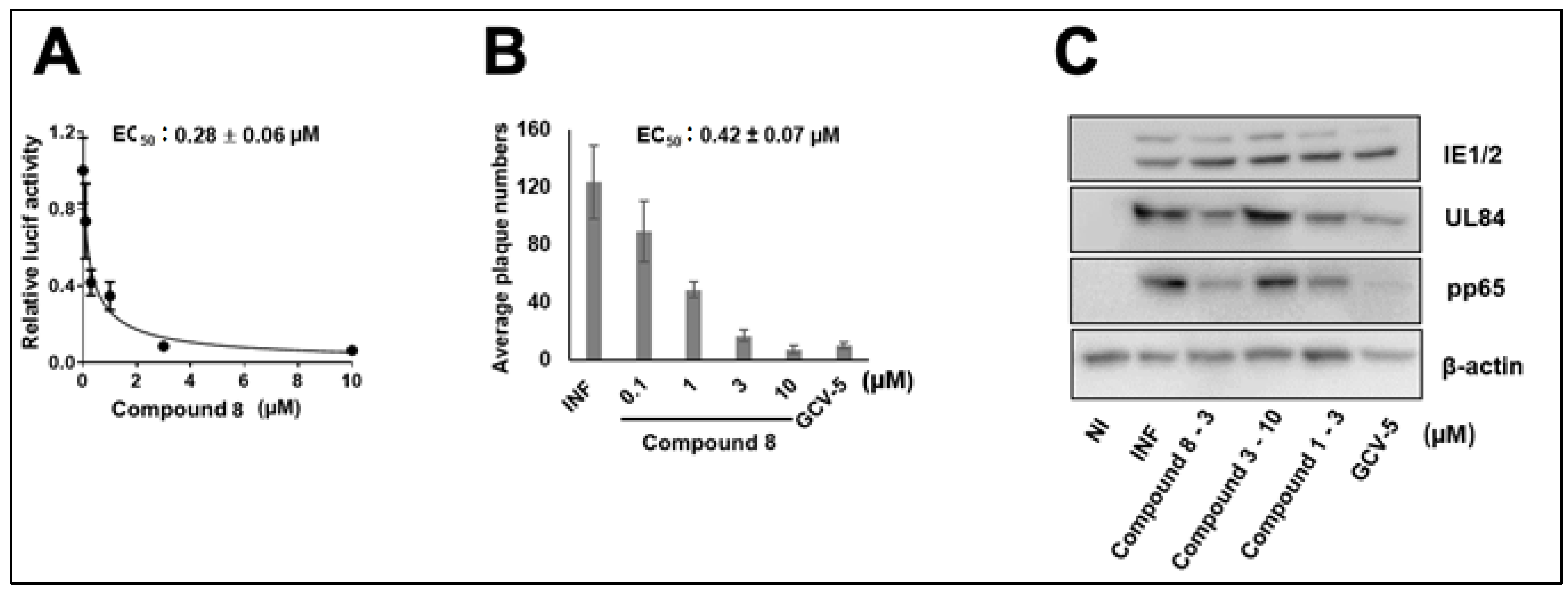
3.1. CMV Inhibition by NCGC2955 Analogs
3.2. CMV Inhibition by Pyridine Analogs of NCGC2955
3.3. NCGC2955 Analogs Inhibit CMV in Primary Human Hepatocytes
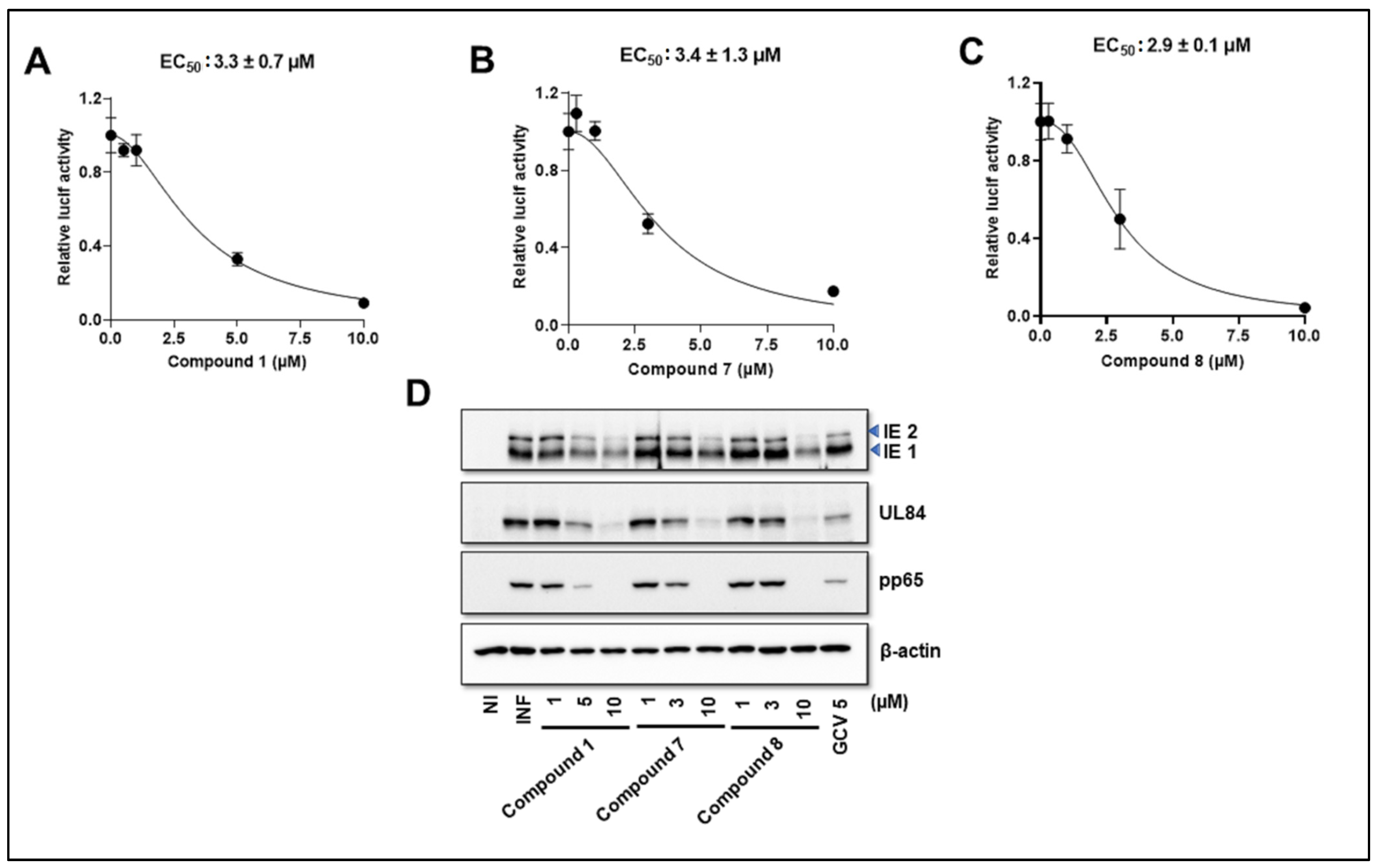
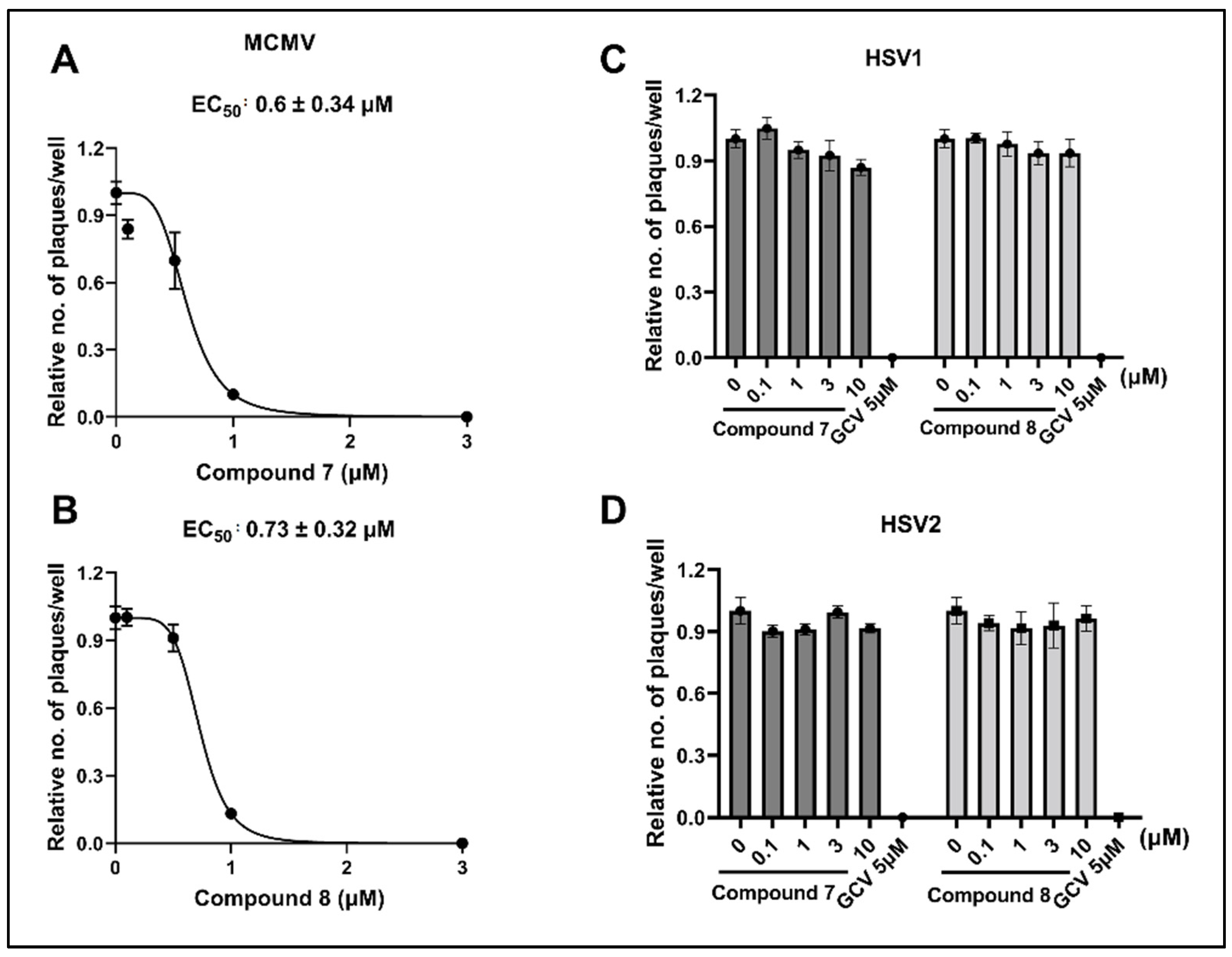
3.4. NCGC2955 Analogs Inhibit Mouse CMV (MCMV) but Not Herpes Simplex Virus 1 or 2 (HSV1 or 2)
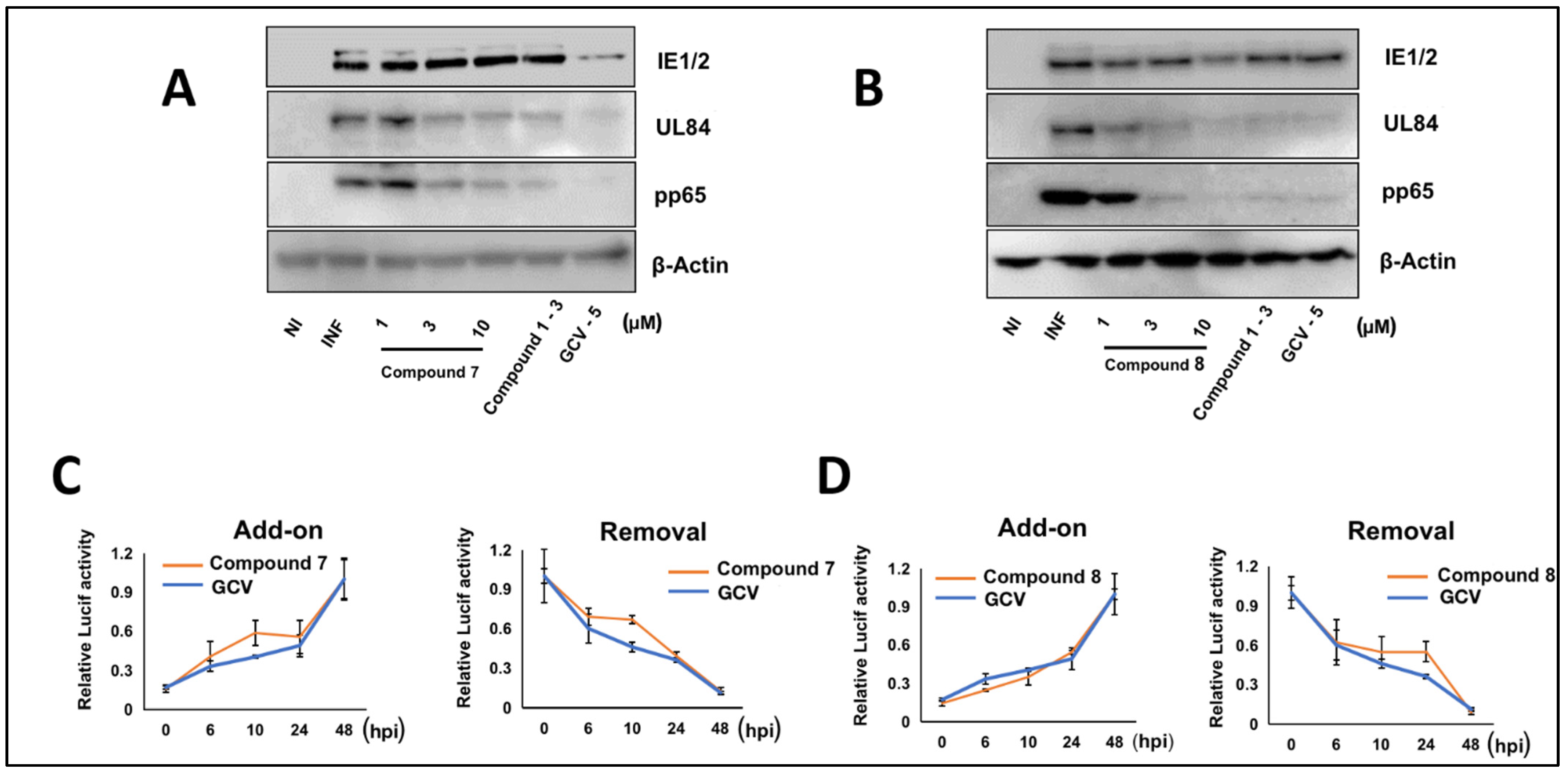
3.5. NCGC2955 Analogs Are Late Inhibitors of CMV Replication
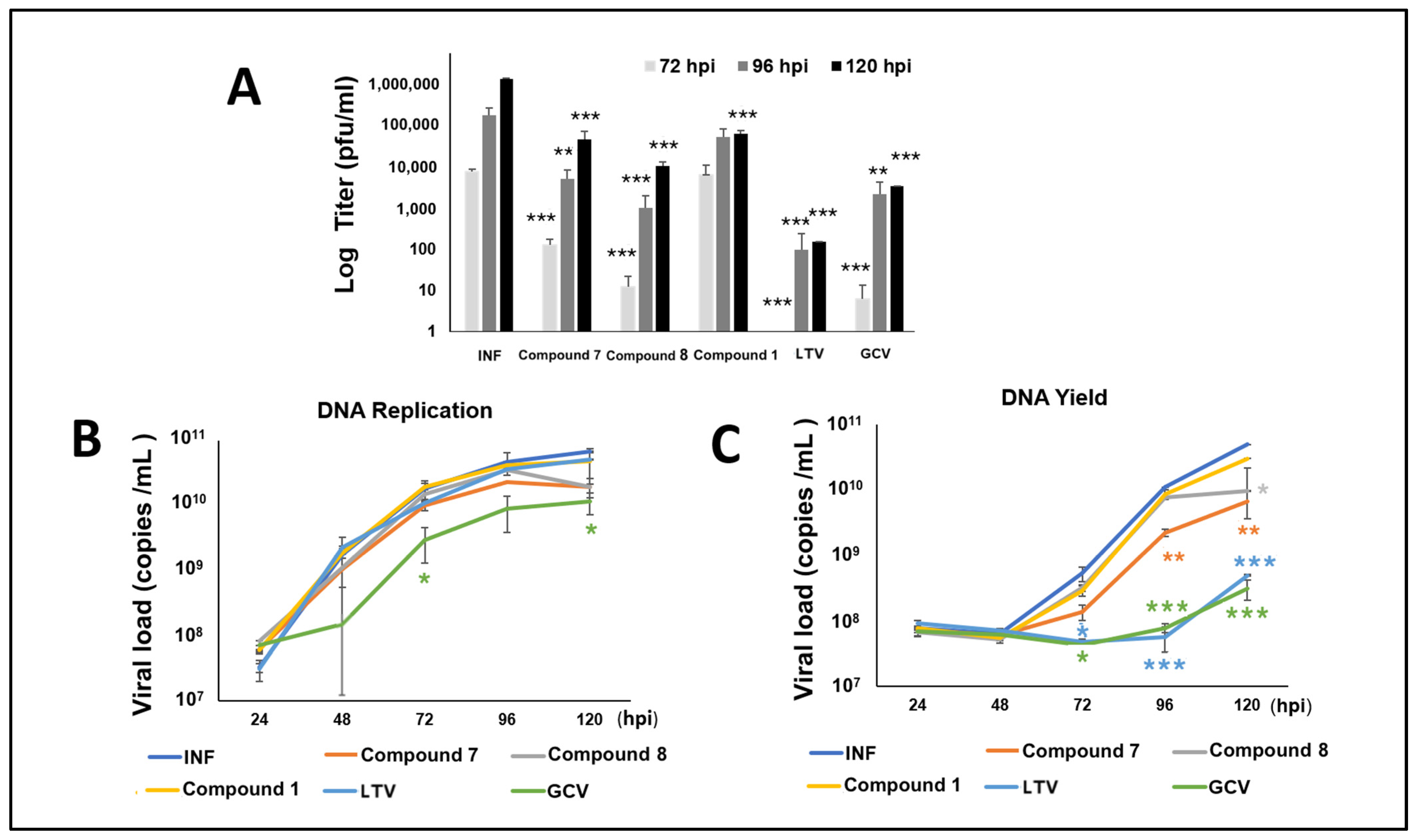
3.6. NCGC2955 Analogs Reduce CMV Yield
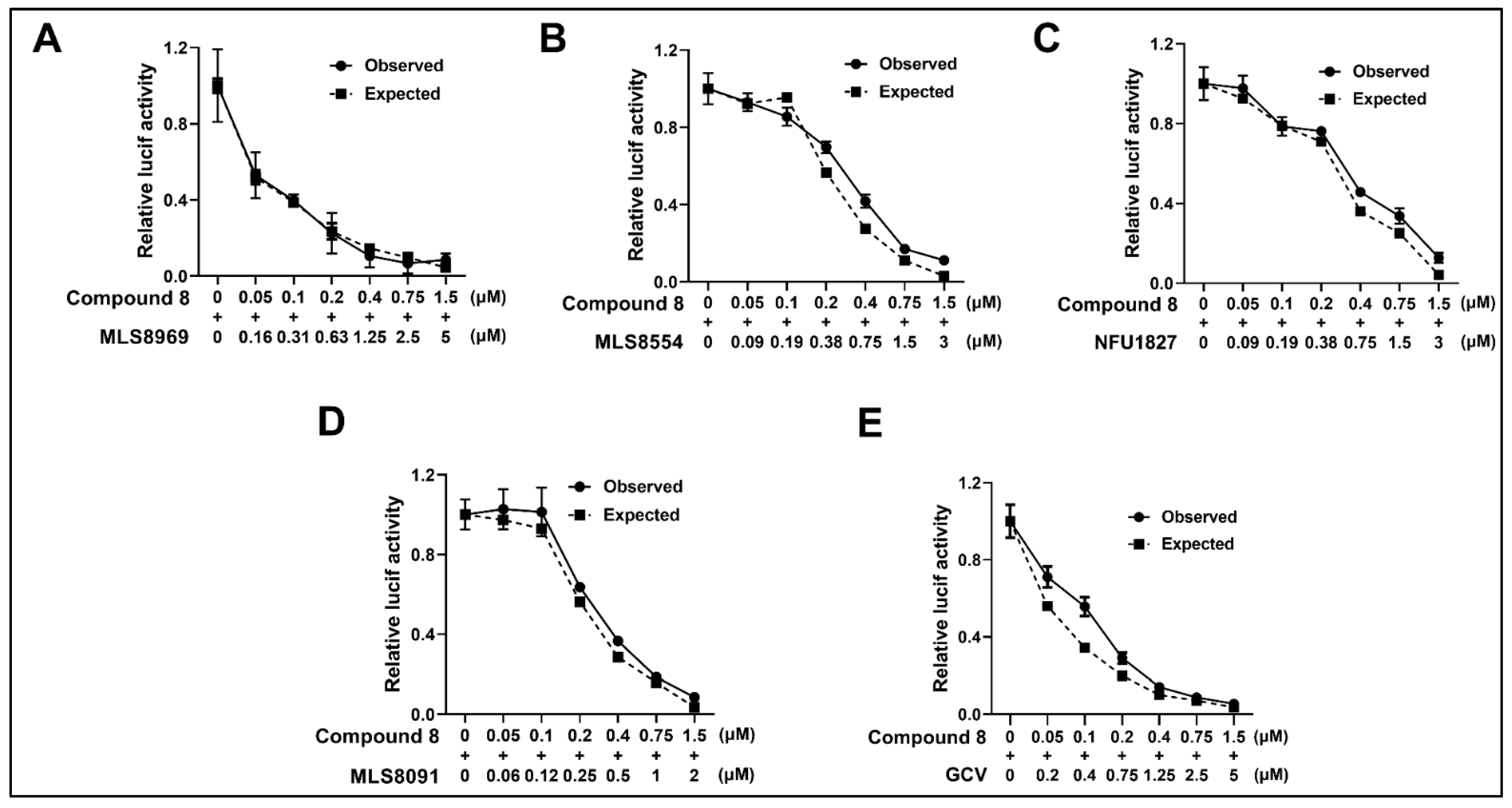
3.7. NCGC2955 Analogs Are Additive with Newly Identified CMV Inhibitors and GCV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Staras, S.A.S.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.; Cannon, M.J. Seroprevalence of Cytomegalovirus Infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Clark, D.A.; Emery, V.C. Betaherpesviruses in transplant recipients. J. Antimicrob. Chemother. 2000, 45, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Jabs, D.A.; Martin, B.K.; Forman, M.S. Mortality Associated with Resistant Cytomegalovirus among Patients with Cytomegalovirus Retinitis and AIDS. Ophthalmology 2010, 117, 128–132.e2. [Google Scholar] [CrossRef] [Green Version]
- Khamduang, W.; Jourdain, G.; Sirirungsi, W.; Layangool, P.; Kanjanavanit, S.; Krittigamas, P.; Pagdi, K.; Somsamai, R.; Sirinontakan, S.; Hinjiranandana, T.; et al. The interrelated transmission of HIV-1 and cytomegalovirus during gestation and delivery in the offspring of HIV-infected mothers. J. Acquir. Immune Defic. Syndr. 2011, 58, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, A.; Schluchter, M.; Easley, K.; Demmler, G.; Shearer, W.; La Russa, P.; Pitt, J.; Cooper, E.; Goldfarb, J.; Hodes, D.; et al. Cytomegalovirus Infection and HIV-1 Disease Progression in Infants Born to HIV-1–Infected Women. N. Engl. J. Med. 1999, 341, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Barbi, M.; Binda, S.; Caroppo, S.; Ambrosetti, U.; Corbetta, C.; Sergi, P. A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr. Infect. Dis. J. 2003, 22, 39–42. [Google Scholar] [CrossRef]
- Boppana, S.B.; Fowler, K.B.; Britt, W.J.; Stagno, S.; Pass, R.F. Symptomatic Congenital Cytomegalovirus Infection in Infants Born to Mothers with Preexisting Immunity to Cytomegalovirus. Pediatrics 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Demmler, G.J. Infectious Diseases Society of America and Centers for Disease Control: Summary of a Workshop on Surveillance for Congenital Cytomegalovirus Disease. Clin. Infect. Dis. 1991, 13, 315–329. [Google Scholar] [CrossRef]
- Avery, R.K.; Arav-Boger, R.; Marr, K.A.; Kraus, E.; Shoham, S.; Lees, L.; Trollinger, B.; Shah, P.; Ambinder, R.; Neofytos, D.; et al. Outcomes in Transplant Recipients Treated with Foscarnet for Ganciclovir-Resistant or Refractory Cytomegalovirus Infection. Transplantation 2016, 100, e74–e80. [Google Scholar] [CrossRef] [Green Version]
- Chou, S. Cytomegalovirus drug resistance and clinical implications. Transpl. Infect. Dis. 2001, 3, 20–24. [Google Scholar] [CrossRef]
- Schreiber, A.; Härter, G.; Schubert, A.; Bunjes, D.; Mertens, T.; Michel, D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin. Pharmacother. 2009, 10, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Steininger, C. Novel Therapies for Cytomegalovirus Disease. Recent Patents Anti-Infect. Drug Discov. 2007, 2, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberlin, D.W.; Lin, C.-Y.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.F.; Kiell, J.M.; et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized, controlled trial. J. Pediatr. 2003, 143, 16–25. [Google Scholar] [CrossRef]
- Kimberlin, D.W.; Jester, P.M.; Sánchez, P.J.; Ahmed, A.; Arav-Boger, R.; Michaels, M.G.; Ashouri, N.; Englund, J.A.; Estrada, B.; Jacobs, R.F.; et al. Valganciclovir for Symptomatic Congenital Cytomegalovirus Disease. N. Engl. J. Med. 2015, 372, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Lanzieri, T.M.; Caviness, A.C.; Blum, P.; Demmler-Harrison, G.; Congenital Cytomegalovirus Longitudinal Study Group. Progressive, Long-Term Hearing Loss in Congenital CMV Disease After Ganciclovir Therapy. J. Pediatric Infect. Dis. Soc. 2021. [Google Scholar] [CrossRef]
- Chou, S. RapidIn VitroEvolution of Human Cytomegalovirus UL56 Mutations That Confer Letermovir Resistance. Antimicrob. Agents Chemother. 2015, 59, 6588–6593. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Ercolani, R.J.; Lanier, E.R. Novel Cytomegalovirus UL54 DNA Polymerase Gene Mutations Selected In Vitro That Confer Brincidofovir Resistance. Antimicrob. Agents Chemother. 2016, 60, 3845–3848. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, R.F.; Ullmann, A.J.; Stoelben, S.; Richard, M.P.; Bornhäuser, M.; Groth, C.; Einsele, H.; Silverman, M.; Mullane, K.M.; Brown, J.; et al. Letermovir for Cytomegalovirus Prophylaxis in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2014, 370, 1781–1789. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Frietsch, J.J.; Michel, D.; Stamminger, T.; Hunstig, F.; Birndt, S.; Schnetzke, U.; Scholl, S.; Hochhaus, A.; Hilgendorf, I. In Vivo Emergence of UL56 C325Y Cytomegalovirus Resistance to Letermovir in a Patient with Acute Myeloid Leukemia after Hematopoietic Cell Transplantation. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, D.J.; Saliba, F.; Blumberg, E.; Abouljoud, M.; Garcia-Diaz, J.B.; Goss, J.A.; Clough, L.; Avery, R.; Limaye, A.P.; Ericzon, B.G.; et al. Efficacy and Safety of Maribavir Dosed at 100 mg Orally Twice Daily for the Prevention of Cytomegalovirus Disease in Liver Transplant Recipients: A Randomized, Double-Blind, Multicenter Controlled Trial. Arab. Archaeol. Epigr. 2012, 12, 3021–3030. [Google Scholar] [CrossRef]
- Winston, D.J.; Young, J.-A.; Pullarkat, V.; Papanicolaou, G.; Vij, R.; Vance, E.; Alangaden, G.J.; Chemaly, R.F.; Petersen, F.; Chao, N.; et al. Maribavir prophylaxis for prevention of cytomegalovirus infection in allogeneic stem cell transplant recipients: A multicenter, randomized, double-blind, placebo-controlled, dose-ranging study. Blood 2008, 111, 5403–5410. [Google Scholar] [CrossRef]
- Papanicolaou, G.A.; Silveira, F.P.; Langston, A.A.; Pereira, M.R.; Avery, R.K.; Uknis, M.; Wijatyk, A.; Wu, J.; Boeckh, M.; Marty, F.; et al. Maribavir for Refractory or Resistant Cytomegalovirus Infections in Hematopoietic-cell or Solid-organ Transplant Recipients: A Randomized, Dose-ranging, Double-blind, Phase 2 Study. Clin. Infect. Dis. 2019, 68, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Ghosh, A.K.; Forman, M.; Hu, X.; Ye, W.; Southall, N.; Marugan, J.J.; Keyes, R.F.; Smith, B.C.; Meyers, D.J.; et al. Validation and Characterization of Five Distinct Novel Inhibitors of Human Cytomegalovirus. J. Med. Chem. 2020, 63, 3896–3907. [Google Scholar] [CrossRef]
- Delekta, P.C.; Dobry, C.J.; Sindac, J.A.; Barraza, S.J.; Blakely, P.K.; Xiang, J.; Kirchhoff, P.D.; Keep, R.F.; Irani, D.N.; Larsen, S.D.; et al. Novel Indole-2-Carboxamide Compounds Are Potent Broad-Spectrum Antivirals Active against Western Equine Encephalitis Virus In Vivo. J. Virol. 2014, 88, 11199–11214. [Google Scholar] [CrossRef] [Green Version]
- Sindac, J.A.; Barraza, S.J.; Dobry, C.J.; Xiang, J.; Blakely, P.K.; Irani, D.N.; Keep, R.F.; Miller, D.J.; Larsen, S.D. Optimization of novel indole-2-carboxamide inhibitors of neurotropic alphavirus replication. J. Med. Chem. 2013, 56, 9222–9241. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Sandford, G.; Hayward, G.S.; Burns, W.H.; Posner, G.H.; Forman, M.; Arav-Boger, R. Recombinant luciferase-expressing human cytomegalovirus (CMV) for evaluation of CMV inhibitors. Virol. J. 2011, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, K.L.; Cavignac, Y.; Stierhof, Y.-D.; Sinzger, C. Human Cytomegalovirus Labeled with Green Fluorescent Protein for Live Analysis of Intracellular Particle Movements. J. Virol. 2005, 79, 2754–2767. [Google Scholar] [CrossRef] [Green Version]
- Forman, M.S.; Vaidya, D.; Bolorunduro, O.; Diener-West, M.; Pass, R.; Arav-Boger, R. Cytomegalovirus Kinetics Following Primary Infection in Healthy Women. J. Infect. Dis. 2017, 215, 1523–1526. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Posner, G.H.; Arav-Boger, R. In VitroCombination of Anti-Cytomegalovirus Compounds Acting through Different Targets: Role of the Slope Parameter and Insights into Mechanisms of Action. Antimicrob. Agents Chemother. 2014, 58, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Ching, K.-C.; Kam, Y.-W.; Merits, A.; Ng, L.F.P.; Chai, C.L.L. Trisubstituted Thieno[3,2-b]pyrrole 5-Carboxamides as Potent Inhibitors of Alphaviruses. J. Med. Chem. 2015, 58, 9196–9213. [Google Scholar] [CrossRef] [PubMed]
- Ching, K.-C.; Tran, T.N.Q.; Amrun, S.N.; Kam, Y.-W.; Ng, L.F.P.; Chai, C.L.L. Structural Optimizations of Thieno[3,2-b]pyrrole Derivatives for the Development of Metabolically Stable Inhibitors of Chikungunya Virus. J. Med. Chem. 2017, 60, 3165–3186. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.E.; Sabalza, M.; Gordts, P.L.S.M.; Spector, D.H. Human Cytomegalovirus Replication Is Inhibited by the Autophagy-Inducing Compounds Trehalose and SMER28 through Distinctively Different Mechanisms. J. Virol. 2018, 92, e02015-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, J.A.; Monk, C.H.; Harrison, M.A.A.; Norton, E.B.; Morris, C.A.; Sullivan, D.E.; Zwezdaryk, K.J. Inhibiting cytomegalovirus replication through targeting the host electron transport chain. Antivir. Res. 2021, 194, 105159. [Google Scholar] [CrossRef]
- Hahn, F.; Niesar, A.; Wangen, C.; Wild, M.; Grau, B.; Herrmann, L.; Capci, A.; Adrait, A.; Couté, Y.; Tsogoeva, S.B.; et al. Target verification of artesunate-related antiviral drugs: Assessing the role of mitochondrial and regulatory proteins by click chemistry and fluorescence labeling. Antivir. Res. 2020, 180, 104861. [Google Scholar] [CrossRef]
- Mercorelli, B.; Luganini, A.; Celegato, M.; Palù, G.; Gribaudo, G.; Lepesheva, G.I.; Loregian, A. The Clinically Approved Antifungal Drug Posaconazole Inhibits Human Cytomegalovirus Replication. Antimicrob. Agents Chemother. 2020, 64, e00056-20. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Venkatadri, R.; Katsnelson, J.; Arav-Boger, R. Digitoxin Suppresses Human Cytomegalovirus Replication via Na(+), K(+)/ATPase alpha1 Subunit-Dependent AMP-Activated Protein Kinase and Autophagy Activation. J. Virol. 2018, 92, e01861-17. [Google Scholar] [CrossRef] [Green Version]
- Sinzger, C.; Bissinger, A.L.; Viebahn, R.; Oettle, H.; Radke, C.; Schmidt, C.A.; Jahn, G. Hepatocytes are Permissive for Human Cytomegalovirus Infection in Human Liver Cell Culture and In Vivo. J. Infect. Dis. 1999, 180, 976–986. [Google Scholar] [CrossRef]
- Chou, S.; Van Wechel, L.C.; Marousek, G.I. Effect of Cell Culture Conditions on the Anticytomegalovirus Activity of Maribavir. Antimicrob. Agents Chemother. 2006, 50, 2557–2559. [Google Scholar] [CrossRef] [Green Version]
- Ligat, G.; Cazal, R.; Hantz, S.; Alain, S. The human cytomegalovirus terminase complex as an antiviral target: A close-up view. FEMS Microbiol. Rev. 2018, 42, 137–145. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | Structure | EC50 pp28-Luciferase | EC50 TB40 | CC50 | Comp | Structure | EC50 pp28-Luciferase | EC50 TB40 | CC50 |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  | 1.7 ± 0.6 | 1.99 ± 0.15 | >200 | 8 |  | 0.28 ± 0.06 | 0.42 ± 0.07 | 500 |
| 2 |  | >30 | 9 |  | 0.3 ± 0.05 | 0.35 ± 0.07 | >500 | ||
| 3 |  | >30 | 10 |  | >30 | ||||
| 4 |  | >30 | 11 |  | >30 | ||||
| 5 |  | >30 | 12 |  | >30 | ||||
| 6 |  | >30 | 13 |  | >30 | ||||
| 7 |  | 0.21 ± 0.06 | 0.55 ± 0.06 | >500 | 14 |  | >30 |
| Compound 1 | EC50 (µM) | Compound 2 | EC50 (µM) | Bliss Coefficient |
|---|---|---|---|---|
| 8 | 0.29 ± 0.1 | MLS8969 | 0.25 ± 0.3 | 1 |
| 8 | 0.41 ± 0.2 | MLS8554 | 0.55 ± 0.2 | 1.2 |
| 8 | 0.35 ± 0.07 | NFU1827 | 0.82 ± 0.1 | 1.2 |
| 8 | 0.27 ± 0.1 | MLS8091 | 0.34 ± 0.1 | 1.1 |
| 8 | 0.38 ± 0.08 | GCV | 0.25 ± 0.1 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Ghosh, A.K.; Keyes, R.F.; Peterson, F.; Forman, M.; Meyers, D.J.; Arav-Boger, R. The Synthesis and Anti-Cytomegalovirus Activity of Piperidine-4-Carboxamides. Viruses 2022, 14, 234. https://doi.org/10.3390/v14020234
Guo X, Ghosh AK, Keyes RF, Peterson F, Forman M, Meyers DJ, Arav-Boger R. The Synthesis and Anti-Cytomegalovirus Activity of Piperidine-4-Carboxamides. Viruses. 2022; 14(2):234. https://doi.org/10.3390/v14020234
Chicago/Turabian StyleGuo, Xin, Ayan Kumar Ghosh, Robert F. Keyes, Francis Peterson, Michael Forman, David J. Meyers, and Ravit Arav-Boger. 2022. "The Synthesis and Anti-Cytomegalovirus Activity of Piperidine-4-Carboxamides" Viruses 14, no. 2: 234. https://doi.org/10.3390/v14020234
APA StyleGuo, X., Ghosh, A. K., Keyes, R. F., Peterson, F., Forman, M., Meyers, D. J., & Arav-Boger, R. (2022). The Synthesis and Anti-Cytomegalovirus Activity of Piperidine-4-Carboxamides. Viruses, 14(2), 234. https://doi.org/10.3390/v14020234