
Next-Generation Sequencing for Confronting Virus Pandemics

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. NGS for Confronting the Devastating Effects of a Pandemic
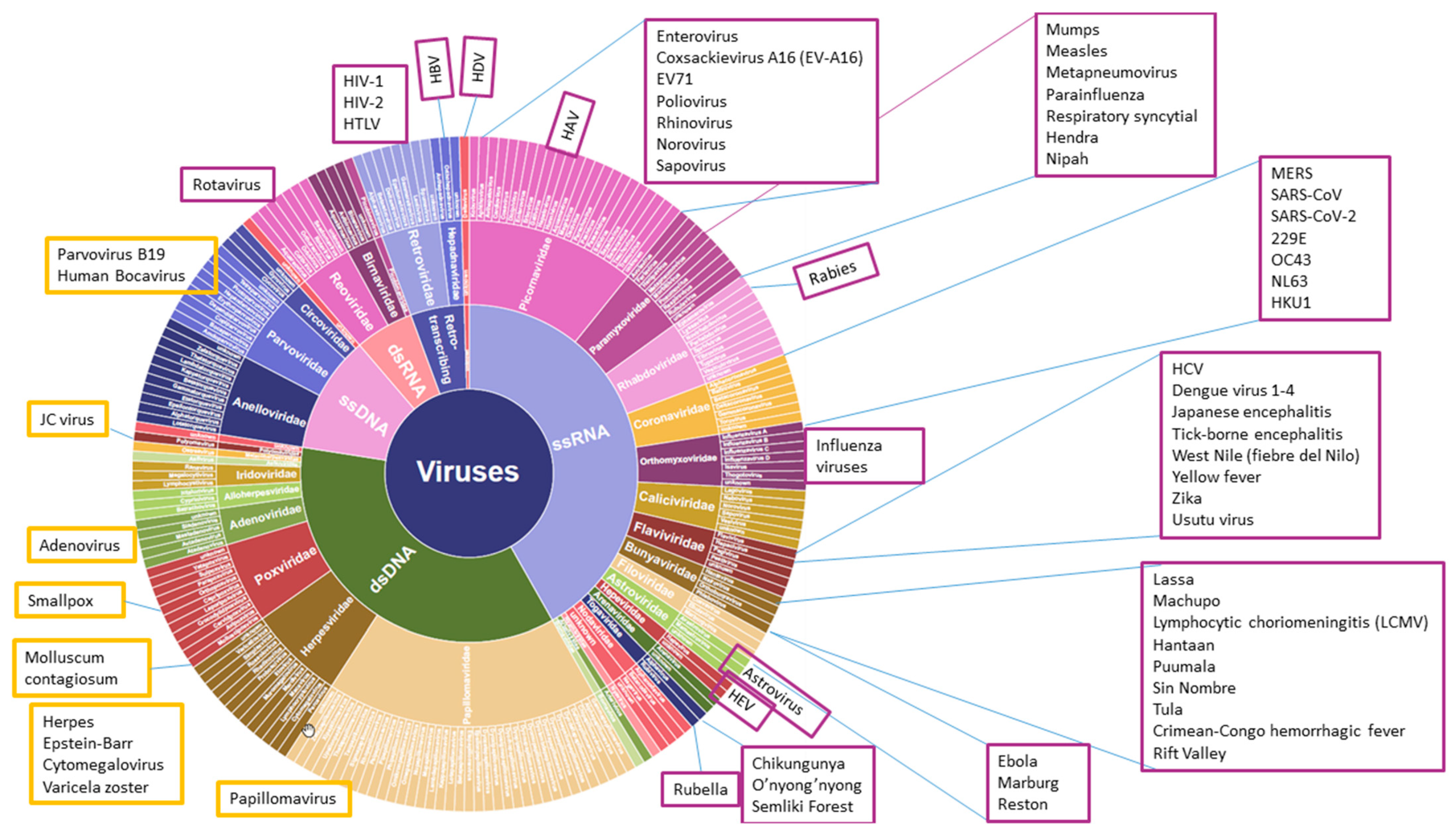
3. Zoonosis and NGS
4. NGS for Studying Genomic Viral Variability
- Pathogenesis or virulence;
- Ability to induce peripheral tolerance = escape from natural immune response;
- Escape from vaccination;
- Escape from drug treatment;
- Changes in tissue or host tropism.
5. NGS as a Diagnostic Tool
6. NGS for Studying the Origin of a Virus and Fighting against Fake Theories
7. NGS Methodologies for Emerging, Re-Emerging, and New Viruses
7.1. NGS Using Random Primers (Metagenomics Sequencing)
- It does not require prior knowledge of the genome for primer or probe design, which simplifies the process overall;
- It allows for the identification of all pathogens in any kind of sample (plasma, serum, feces, cerebrospinal fluid, sewage, etc.) as well as the major genomes of the viral population in epidemiological studies, or outbreak investigations.
7.2. Sequence Capture, Fragment Recovery and NGS Sequencing (Target Enrichment Sequencing)
- The sequence capture strategy allows for enrichment of sequences from specific microorganisms in a complex mixture;
- Whole genomes may be sequenced at a low cost, as non-pathogenic genomes are significantly reduced, thus reducing the contaminating nucleic acids from different origins. Thanks to this, few PCR cycles are necessary for amplification, limiting the introduction of artefact mutations, and minor variants are preserved (i.e., higher sensitivity), reflecting in vivo variation.
- The design of oligonucleotide capture probes requires knowledge of the microorganism genome. Thus, it does not allow for the sequencing of novel pathogens;
- It requires high technical expertise for sample pre-treatment;
- Although it is highly sensitive, the recovered sequencing coverage is low.
7.3. Use of Amplicons (PCR Amplicon Sequencing)
- It is a highly sensitive and specific method, widely used, with trusted and well-established methods;
- It is able to specifically amplify the infectious agents, significantly decreasing the non-pathogen genomic background, and increasing cost-effectiveness per sample.
- It achieves good coverage even at a low pathogen load;
- It is the most sensitive method for genome sequencing from a complex mixture, thus, allowing for the detection of variants at very low frequencies.
- It is highly dependent on the primer’s quality, specificity, and mismatch, particularly in poorly characterized pathogens, in pathogens with recognized high genetic diversity or those with novel variants;
- The higher the number of cycles, the higher the number of artefact mutations;
- It requires knowledge of the microorganism genome to design specific primers.
7.4. Next-Next Generation Sequencing (NNGS) or Third Generation Long-Read Sequencing (TGS)
8. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hwang, J.Y.; Ahn, S.J.; Kwon, M.-G.; Seo, J.S.; Hwang, S.D.; Jee, B.Y. Whole-genome next-generation sequencing and phylogenetic characterization of viral haemorrhagic septicaemia virus in Korea. J. Fish Dis. 2020, 43, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yll, M.; Cortese, M.F.; Guerrero-Murillo, M.; Orriols, G.; Gregori, J.; Casillas, R.; González, C.; Sopena, S.; Godoy, C.; Vila, M.; et al. Conservation and variability of hepatitis B core at different chronic hepatitis stages. World J. Gastroenterol. 2020, 26, 2584–2598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Perales, C.; Soria, M.E.; García-Cehic, D.; Gregori, J.; Rodríguez-Frías, F.; Buti, M.; Crespo, J.; Calleja, J.L.; Tabernero, D.; et al. Deep-sequencing reveals broad subtype-specific HCV resistance mutations associated with treatment failure. Antiviral Res. 2020, 174, 104694. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef] [PubMed]
- Borah, P.; Deb, P.K.; Al-Shar’i, N.A.; Dahabiyeh, L.A.; Venugopala, K.N.; Singh, V.; Shinu, P.; Hussain, S.; Deka, S.; Chandrasekaran, B.; et al. Perspectives on RNA Vaccine Candidates for COVID-19. Front. Mol. Biosci. 2021, 8, 635245. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Ríos, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.; Piñana, M.; Borràs-Bermejo, B.; González-Sánchez, A.; García-Cehic, D.; Esperalba, J.; Rando, A.; Zules-Oña, R.-G.; Campos, C.; Codina, M.G.; et al. A year living with SARS-CoV-2: An epidemiological overview of viral lineage circulation by whole-genome sequencing in Barcelona city (Catalonia, Spain). Emerg. Microbes Infect. 2022, 11, 172–181. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Edridge, A.W.D.; Deijs, M.; van Zeggeren, I.E.; Kinsella, C.M.; Jebbink, M.F.; Bakker, M.; van de Beek, D.; Brouwer, M.C.; van der Hoek, L. Viral Metagenomics on Cerebrospinal Fluid. Genes 2019, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Glaser, C.A.; Honarmand, S.; Anderson, L.J.; Schnurr, D.P.; Forghani, B.; Cossen, C.K.; Schuster, F.L.; Christie, L.J.; Tureen, J.H. Beyond viruses: Clinical profiles and etiologies associated with encephalitis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2006, 43, 1565–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailles, A.; Stahl, J.-P. Infectious encephalitis in france in 2007: A national prospective study. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 49, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Studahl, M.; Lindquist, L.; Eriksson, B.-M.; Günther, G.; Bengner, M.; Franzen-Röhl, E.; Fohlman, J.; Bergström, T.; Aurelius, E. Acute viral infections of the central nervous system in immunocompetent adults: Diagnosis and management. Drugs 2013, 73, 131–158. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cassi, X.; Martínez-Puchol, S.; Silva-Sales, M.; Cornejo, T.; Bartolome, R.; Bofill-Mas, S.; Girones, R. Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach. Viruses 2020, 12, 1432. [Google Scholar] [CrossRef] [PubMed]
- Racsa, L.D.; Kraft, C.S.; Olinger, G.G.; Hensley, L.E. Viral Hemorrhagic Fever Diagnostics. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2016, 62, 214–219. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, C.J.; Sawyer, S.L. How host genetics dictates successful viral zoonosis. PLoS Biol. 2019, 17, e3000217. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Barciela, M.; Minguez, B.; Girones, R.; Rodriguez-Frias, F.; Quer, J.; Buti, M. Phylogenetic demonstration of hepatitis E infection transmitted by pork meat ingestion. J. Clin. Gastroenterol. 2015, 49, 165–168. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Reinfection with SARS-CoV-2: Implementation of a Surveillance Case Definition within the EU/EEA; ECDC: Stockholm, Sweden, 2021. [Google Scholar]
- Hampson, A.; Barr, I.; Cox, N.; Donis, R.O.; Siddhivinayak, H.; Jernigan, D.; Katz, J.; McCauley, J.; Motta, F.; Odagiri, T.; et al. Improving the selection and development of influenza vaccine viruses - Report of a WHO informal consultation on improving influenza vaccine virus selection, Hong Kong SAR, China, 18-20 November 2015. Vaccine 2017, 35, 1104–1109. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Saelens, X.; Thomas, I.; Roosens, N.H. Next-Generation Sequencing: An Eye-Opener for the Surveillance of Antiviral Resistance in Influenza. Trends Biotechnol. 2020, 38, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilden, D. Hilary Koprowski, MD. December 5, 1916-April 11, 2013. J. Neurovirol. 2013, 19, 195–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabin, A.B. Strategy for rapid elimination and continuing control of poliomyelitis and other vaccine preventable diseases of children in developing countries. Br. Med. J. 1986, 292, 531–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Global Polio Eradication Initiative Applauds Who African Region for Wild Polio-Free Certification; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Jorgensen, D.; Pons-Salort, M.; Shaw, A.G.; Grassly, N.C. The role of genetic sequencing and analysis in the polio eradication programme. Virus Evol. 2020, 6, veaa040. [Google Scholar] [CrossRef] [PubMed]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular Evidence of Sexual Transmission of Ebola Virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lednicky, J.; Beau De Rochars, V.M.; El Badry, M.; Loeb, J.; Telisma, T.; Chavannes, S.; Anilis, G.; Cella, E.; Ciccozzi, M.; Rashid, M.; et al. Zika Virus Outbreak in Haiti in 2014: Molecular and Clinical Data. PLoS Negl. Trop. Dis. 2016, 10, e0004687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrerizo, M.; García-Iñiguez, J.P.; Munell, F.; Amado, A.; Madurga-Revilla, P.; Rodrigo, C.; Pérez, S.; Martínez-Sapiña, A.; Antón, A.; Suárez, G.; et al. First Cases of Severe Flaccid Paralysis Associated With Enterovirus D68 Infection in Spain, 2015-2016. Pediatr. Infect. Dis. J. 2017, 36, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Wang, J.; Wang, J.; Lu, Y.; Zhang, Y.; Peng, R.; Lu, J.; Chen, Z. The Emergence and Spread of Novel SARS-CoV-2 Variants. Front. Public Health 2021, 9, 1017. [Google Scholar] [CrossRef] [PubMed]
- González-Candelas, F.; Shaw, M.-A.; Phan, T.; Kulkarni-Kale, U.; Paraskevis, D.; Luciani, F.; Kimura, H.; Sironi, M. One year into the pandemic: Short-term evolution of SARS-CoV-2 and emergence of new lineages. Infect. Genet. Evol. 2021, 92, 104869. [Google Scholar] [CrossRef]
- Piñana, M.; Abril, J.F.; Andrés, C.; Silgado, A.; Navarro, A.; Suy, A.; Sulleiro, E.; Pumarola, T.; Quer, J.; Antón, A. Viral populations of SARS-CoV-2 in upper respiratory tract, placenta, amniotic fluid and umbilical cord blood support viral replication in placenta. Clin. Microbiol. Infect. 2021, 27, 1542–1544. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Heavily mutated Omicron variant puts scientists on alert. Nature 2021, 600, 21. [Google Scholar] [CrossRef] [PubMed]
- World Health Organizaton. Draft Landscape and Tracker of COVID-19 Candidate Vaccines; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Kim, Y.C.; Dema, B.; Reyes-Sandoval, A. COVID-19 vaccines: Breaking record times to first-in-human trials. NPJ Vaccines 2020, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.W. The Health 202: The FDA Isn’t Slowing the Development of a Coronavirus Vaccine as Trump Claims. Available online: https://www.washingtonpost.com/politics/2020/08/25/health-202-fda-isnt-slowing-development-coronavirus-vaccine-trump-claims/ (accessed on 4 February 2022).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John Hopkins University (JHU). COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at John Hopkins University (JHU); Johns Hopkins University & Medicine: Baltimore, MA, USA, 2022; Available online: https://coronavirus.jhu.edu/map.html (accessed on 4 February 2022).
- Josh Quick, N.L. ARTIC Network. 2019. Available online: https://artic.network/ncov-2019 (accessed on 4 February 2022).
- Kupferschmidt, K. Where did ‘weird’ Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Emegencies. Disease Outbreaks. Pandemic, Epidemic Diseases; WHO: Geneva, Switzerland, 2022; Available online: https://www.who.int/emergencies/diseases/en/ (accessed on 4 February 2022).
- Dimaio, D. Is virology dead? MBio 2014, 5, e01003-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. Zoonotic Diseases. 2021. Available online: https://www.cdc.gov/onehealth/basics/zoonotic-diseases.html (accessed on 4 February 2022).
- Hawkins, J.A.; Kaczmarek, M.E.; Müller, M.A.; Drosten, C.; Press, W.H.; Sawyer, S.L. A metaanalysis of bat phylogenetics and positive selection based on genomes and transcriptomes from 18 species. Proc. Natl. Acad. Sci. USA 2019, 116, 11351–11360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Sole, R.V.; Ferrer, R.; Gonzalez-Garcia, I.; Quer, J.; Domingo, E. Red queen dynamics, competition and critical points in a model of RNA virus quasispecies. J. Theor. Biol. 1999, 198, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quer, J.; Huerta, R.; Novella, I.S.; Tsimring, L.; Domingo, E.; Holland, J.J. Reproducible nonlinear population dynamics and critical points during replicative competitions of RNA virus quasispecies. J. Mol. Biol. 1996, 264, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; Escarmis, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, R.L. Viruses as an etiology of obesity. Mayo Clin. Proc. 2007, 82, 1192–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, V.J.; deJager, S.A.; Grauls, G.E.; Gielen, M.; Vlietinck, R.F.; Derom, C.A.; Loos, R.J.F.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; et al. Lack of evidence for the role of human adenovirus-36 in obesity in a European cohort. Obesity 2011, 19, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Na, H.-N.; Nam, J.-H. Adenovirus 36 as an obesity agent maintains the obesity state by increasing MCP-1 and inducing inflammation. J. Infect. Dis. 2012, 205, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Woolford, L.; Rector, A.; Van Ranst, M.; Ducki, A.; Bennett, M.D.; Nicholls, P.K.; Warren, K.S.; Swan, R.A.; Wilcox, G.E.; O’Hara, A.J. A novel virus detected in papillomas and carcinomas of the endangered western barred bandicoot (Perameles bougainville) exhibits genomic features of both the Papillomaviridae and Polyomaviridae. J. Virol. 2007, 81, 13280–13290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; Alhakeem, R.; Durosinloun, A.; Al Asmari, M.; Islam, A.; et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasso, G.; Mayer, S.V.; Winkelmann, E.R.; Chu, T.; Elliot, O.; Patino-Galindo, J.A.; Park, K.; Rabadan, R.; Honig, B.; Shapira, S.D. A Structure-Informed Atlas of Human-Virus Interactions. Cell 2019, 178, 1526–1541.e16. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Ali Khan, S.; et al. A strategy to estimate unknown viral diversity in mammals. MBio 2013, 4, e00598-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Sikkema, R.S.; Koopmans, M.P.G. Preparing for Emerging Zoonotic Viruses. Encycl. Virol. 2021, 256–266. [Google Scholar] [CrossRef]
- Ganem, D.; Neill, U.S. A conversation with Don Ganem. J. Clin. Invest. 2014, 124, 464–465. [Google Scholar] [PubMed] [Green Version]
- Perales, C.; Chen, Q.; Soria, M.E.; Gregori, J.; Garcia-Cehic, D.; Nieto-Aponte, L.; Castells, L.; Imaz, A.; Llorens-Revull, M.; Domingo, E.; et al. Baseline hepatitis C virus resistance-associated substitutions present at frequencies lower than 15% may be clinically significant. Infect. Drug Resist. 2018, 11, 2207–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekse, C.; Holst-Jensen, A.; Dobrindt, U.; Johannessen, G.S.; Li, W.; Spilsberg, B.; Shi, J. High Throughput Sequencing for Detection of Foodborne Pathogens. Front. Microbiol. 2017, 8, 2029. [Google Scholar] [CrossRef] [PubMed]
- Brunker, K.; Nadin-Davis, S.; Biek, R. Genomic sequencing, evolution and molecular epidemiology of rabies virus. Rev. Sci. Tech. 2018, 37, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- McGinnis, J.; Laplante, J.; Shudt, M.; George, K.S. Next generation sequencing for whole genome analysis and surveillance of influenza A viruses. J. Clin. Virol. 2016, 79, 44–50. [Google Scholar] [CrossRef]
- Domingo, E. Virus as Populations. Composition, Complexity, Dynamics and Biological Implications. In Virus as Populations, Vol. First ed.; Academic Press, Elsevier: London, UK, 2016; pp. 1–412. [Google Scholar]
- Bartenschlager, R.; Lohmann, V. Replication of hepatitis C virus. J. Gen. Virol. 2000, 81, 1631–1648. [Google Scholar] [PubMed]
- Herrmann, E.; Neumann, A.U.; Schmidt, J.M.; Zeuzem, S. Hepatitis C virus kinetics. Antivir. Ther. 2000, 5, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.P.; Neumann, A.U.; Gretch, D.R.; Wiley, T.E.; Perelson, A.S.; Layden, T.J. Dose-dependent acute clearance of hepatitis C genotype 1 virus with interferon alfa. Hepatology 1997, 26, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Gaudieri, S.; Rauch, A.; Pfafferott, K.; Barnes, E.; Cheng, W.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; Mollison, L.; Baker, R.; et al. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2009, 49, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; James, I.; Pfafferott, K.; Nolan, D.; Klenerman, P.; Cheng, W.; Mollison, L.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; et al. Divergent adaptation of hepatitis C virus genotypes 1 and 3 to human leukocyte antigen-restricted immune pressure. Hepatology 2009, 50, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. Mutation rates and rapid evolution of RNA viruses. In Evolutionary Biology of Viruses; Morse, S.S., Ed.; Raven Press: New York, NY, USA, 1994; pp. 161–184. [Google Scholar]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Martell, M.; Esteban, J.I.; Quer, J.; Genesc, J.; Weiner, A.; Esteban, R.; Guardia, J.; G?mez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, J.J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. High error rates, population equilibrium, and evolution of RNA replication systems. In RNA Genetics; Domingo, E., Holland, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 1988; pp. 3–36. [Google Scholar]
- Rodriguez-Frias, F.; Nieto-Aponte, L.; Gregori, J.; Garcia-Cehic, D.; Casillas, R.; Tabernero, D.; Homs, M.; Blasi, M.; Vila, M.; Chen, Q.; et al. High HCV subtype heterogeneity in a chronically infected general population revealed by high-resolution hepatitis C virus subtyping. Clin. Microbiol. Infect. 2017, 23, 775.e1–775.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quer, J.; Gregori, J.; Rodriguez-Frias, F.; Buti, M.; Madejon, A.; Perez-del-Pulgar, S.; Garcia-Cehic, D.; Casillas, R.; Blasi, M.; Homs, M.; et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J. Clin. Microbiol. 2015, 53, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.C.; Denison, M.R. Coronaviruses as DNA wannabes: A new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013, 9, e1003760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, J.; Pruijssers, A.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Stevens, L.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.J. Replication Error, Quasispecies Populations and Extreme Evolution Rates of RNA Viruses; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, Á.; Scher, E.; Rambaut, A. SARS-CoV-2. Lineages. PANGOLIN. 2022. Available online: https://cov-lineages.org/resources/pangolin.html (accessed on 4 February 2022).
- Sender, R.; Bar-On, Y.M.; Gleizer, S.; Bernshtein, B.; Flamholz, A.; Phillips, R.; Milo, R. The total number and mass of SARS-CoV-2 virions. Proc. Natl. Acad. Sci. USA 2021, 118, e2024815118. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19 - Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A. Omicron emerges. New Sci. 2021, 252, 7. [Google Scholar] [CrossRef]
- Rowe, C.L.; Fleming, J.O.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Baker, S.C. Generation of coronavirus spike deletion variants by high-frequency recombination at regions of predicted RNA secondary structure. J. Virol. 1997, 71, 6183–6190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, C.L.; Baker, S.C.; Nathan, M.J.; Fleming, J.O. Evolution of mouse hepatitis virus: Detection and characterization of spike deletion variants during persistent infection. J. Virol. 1997, 71, 2959–2969. [Google Scholar] [CrossRef] [Green Version]
- Muth, D.; Corman, V.M.; Roth, H.; Binger, T.; Dijkman, R.; Gottula, L.T.; Gloza-Rausch, F.; Balboni, A.; Battilani, M.; Rihtaric, D.; et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 2018, 8, 15177. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-Y.; Wang, P.; Mok, B.W.-Y.; Zhang, A.J.; Chu, H.; Lee, A.C.-Y.; Deng, S.; Chen, P.; Chan, K.-H.; Song, W.; et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 2020, 9, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Lau, S.Y.; Wang To, K.K.; Mok, B.W.Y.; Li, X.; Wang, P.; Deng, S.; Woo, K.F.; Du, Z.; Li, C.; et al. Natural Transmission of Bat-like Severe Acute Respiratory Syndrome Coronavirus 2 Without Proline-Arginine-Arginine-Alanine Variants in Coronavirus Disease 2019 Patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 73, e437–e444. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Brandt, D.; Simunovic, M.; Busche, T.; Haak, M.; Belmann, P.; Jünemann, S.; Schulz, T.; Klages, L.J.; Vinke, S.; Beckstette, M.; et al. Multiple Occurrences of a 168-Nucleotide Deletion in SARS-CoV-2 ORF8, Unnoticed by Standard Amplicon Sequencing and Variant Calling Pipelines. Viruses 2021, 13, 1870. [Google Scholar] [CrossRef] [PubMed]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef] [PubMed]
- Moldován, N.; Szucs, A.; Tombácz, D.; Balázs, Z.; Csabai, Z.; Snyder, M.; Boldogkoi, Z. Multiplatform next-generation sequencing identifies novel RNA molecules and transcript isoforms of the endogenous retrovirus isolated from cultured cells. FEMS Microbiol. Lett. 2018, 365, fny013. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kao, C. Factors regulating template switch in vitro by viral RNA-dependent RNA polymerases: Implications for RNA-RNA recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 4972–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.D.; Simon, A.E. New insights into the mechanisms of RNA recombination. Virology 1997, 235, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.A.; Morris, T.J. Defective and defective interfering RNAs of monopartite plus-strand RNA plant viruses. Curr. Top. Microbiol. Immunol. 1999, 239, 1–17. [Google Scholar] [CrossRef]
- DePolo, N.J.; Giachetti, C.; Holland, J.J. Continuing coevolution of virus and defective interfering particles and of viral genome sequences during undiluted passages: Virus mutants exhibiting nearly complete resistance to formerly dominant defective interfering particles. J. Virol. 1987, 61, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachetti, C.; Holland, J.J. Altered replicase specificity is responsible for resistance to defective interfering particle interference of an Sdi- mutant of vesicular stomatitis virus. J. Virol. 1988, 62, 3614–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Magnus, P. Incomplete forms of influenza virus. Adv. Virus Res. 1954, 2, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Moreno, I.; Sanjuán, R. Collective Viral Spread Mediated by Virion Aggregates Promotes the Evolution of Defective Interfering Particles. MBio 2020, 11, e02156-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasiów-Jaroszewska, B.; Minicka, J.; Zarzyńska-Nowak, A.; Budzyńska, D.; Elena, S.F. Defective RNA particles derived from Tomato black ring virus genome interfere with the replication of parental virus. Virus Res. 2018, 250, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, T.-Y.; Liao, W.-Y.; Wu, H.-Y. A leaderless genome identified during persistent bovine coronavirus infection is associated with attenuation of gene expression. PLoS ONE 2013, 8, e82176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemsen, A.; Carrasco, J.L.; Elena, S.F.; Zwart, M.P. Going, going, gone: Predicting the fate of genomic insertions in plant RNA viruses. Heredity 2018, 121, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Sardanyés, J.; Elena, S.F. Error threshold in RNA quasispecies models with complementation. J. Theor. Biol. 2010, 265, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, S.; Galeano, F.; Locatelli, F.; Gallo, A. ADARs and the Balance Game between Virus Infection and Innate Immune Cell Response. Curr. Issues Mol. Biol. 2015, 17, 37–51. [Google Scholar]
- Cattaneo, R. Biased (A-->I) hypermutation of animal RNA virus genomes. Curr. Opin. Genet. Dev. 1994, 4, 895–900. [Google Scholar] [CrossRef]
- Phuphuakrat, A.; Kraiwong, R.; Boonarkart, C.; Lauhakirti, D.; Lee, T.-H.; Auewarakul, P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J. Virol. 2008, 82, 10864–10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-D.; Na, L.; Fu, L.-H.; Yang, F.; Zhu, C.-H.; Tang, L.; Li, Q.; Wang, J.-Y.; Li, Z.; Wang, X.-F.; et al. Double-stranded RNA-specific adenosine deaminase 1 (ADAR1) promotes EIAV replication and infectivity. Virology 2015, 476, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria, M.; Neri, F.; Gallo, A.; Farace, M.G.; Michienzi, A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res. 2009, 37, 5848–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, C.X.; John, L.; Samuel, C.E. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interf. Cytokine Res. 2014, 34, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L. Control of ADAR1 editing of hepatitis delta virus RNAs. Curr. Top. Microbiol. Immunol. 2012, 353, 123–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregori, J.; Cortese, M.F.; Piñana, M.; Campos, C.; Garcia-Cehic, D.; Andrés, C.; Abril, J.F.; Codina, M.G.; Rando, A.; Esperalba, J.; et al. Host-dependent editing of SARS-CoV-2 in COVID-19 patients. Emerg. Microbes Infect. 2021, 10, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Jerome, H.; Taylor, C.; Sreenu, V.B.; Klymenko, T.; Filipe, A.D.S.; Jackson, C.; Davis, C.; Ashraf, S.; Wilson-Davies, E.; Jesudason, N.; et al. Metagenomic next-generation sequencing aids the diagnosis of viral infections in febrile returning travellers. J. Infect. 2019, 79, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prachayangprecha, S.; Schapendonk, C.M.E.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C.; Pas, S.D.; van der Eijk, A.A.; Poovorawan, Y.; Haagmans, B.L.; Smits, S.L. Exploring the potential of next-generation sequencing in detection of respiratory viruses. J. Clin. Microbiol. 2014, 52, 3722–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bart, G.; Fischer, D.; Samoylenko, A.; Zhyvolozhnyi, A.; Stehantsev, P.; Miinalainen, I.; Kaakinen, M.; Nurmi, T.; Singh, P.; Kosamo, S.; et al. Characterization of nucleic acids from extracellular vesicle-enriched human sweat. BMC Genomics 2021, 22, 425. [Google Scholar] [CrossRef]
- Cho, S.; Kim, W.-K.; No, J.S.; Lee, S.-H.; Jung, J.; Yi, Y.; Park, H.C.; Lee, G.-Y.; Park, K.; Kim, J.-A.; et al. Urinary genome detection and tracking of Hantaan virus from hemorrhagic fever with renal syndrome patients using multiplex PCR-based next-generation sequencing. PLoS Negl. Trop. Dis. 2021, 15, e0009707. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau-Expósito, J.; Perea, D.; Suppi, M.; Massana, N.; Vergara, A.; Soler, M.J.; Trinite, B.; Blanco, J.; García-Pérez, J.; Alcamí, J.; et al. Evaluation of SARS-CoV-2 entry, inflammation and new therapeutics in human lung tissue cells. PLoS Pathog. 2022, 18, e1010171. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buti, M.; Tabernero, D.; Mas, A.; Homs, M.; Prieto, M.; Rodríguez-Frías, F.; Casafont, F.; Casillas, R.; González, A.; Miras, M.; et al. Hepatitis B virus quasispecies evolution after liver transplantation in patients under long-term lamivudine prophylaxis with or without hepatitis B immune globulin. Transpl. Infect. Dis. 2015, 17, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Quer, J.; Rodriguez-Frias, F.; Gregori, J.; Tabernero, D.; Eugenia, S.M.; Garcia-Cehic, D.; Homs, M.; Bosch, A.; Pinto, R.M.; Esteban, J.I.; et al. Deep sequencing in the management of hepatitis virus infections. Virus Res. 2017, 239, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C Virus into 7 genotypes and 67 Subtypes: Updated criteria and assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a Novel Hepatitis C Virus Genotype From Punjab, India: Expanding Classification of Hepatitis C Virus Into 8 Genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiken, C.; Combet, C.; Bukh, J.; Shin, I.; Deleage, G.; Mizokami, M.; Richardson, R.; Sablon, E.; Yusim, K.; Pawlotsky, J.M.; et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 2006, 44, 1355–1361. [Google Scholar] [CrossRef]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef]
- Garcia-Cehic, D.; Rando, A.; Rodriguez-Frias, F.; Gregori, J.; Costa, J.G.; Carrión, J.A.; Macenlle, R.; Pamplona, J.; Castro-Iglesias, A.; Cañizares, A.; et al. Resistance-associated substitutions after sofosbuvir/velpatasvir/voxilaprevir triple therapy failure. J. Viral Hepat. 2021, 28, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Ordeig, L.; Garcia-Cehic, D.; Gregori, J.; Soria, M.E.; Nieto-Aponte, L.; Perales, C.; Llorens, M.; Chen, Q.; Riveiro-Barciela, M.; Buti, M.; et al. New hepatitis C virus genotype 1 subtype naturally harbouring resistance-associated mutations to NS5A inhibitors. J. Gen. Virol. 2018, 99, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health, O. WHO-Convened Global Study of Origins of SARS-CoV-2: China Part; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Calisher, C.H.; Carroll, D.; Colwell, R.; Corley, R.B.; Daszak, P.; Drosten, C.; Enjuanes, L.; Farrar, J.; Field, H.; Golding, J.; et al. Science, not speculation, is essential to determine how SARS-CoV-2 reached humans. Lancet 2021, 398, 209–211. [Google Scholar] [CrossRef]
- Wong, Y.C.; Lau, S.Y.; Wang To, K.K.; Mok, B.W.Y.; Li, X.; Wang, P.; Deng, S.; Woo, K.F.; Du, Z.; Li, C.; et al. Natural transmission of bat-like SARS-CoV-2 deltaPRRA variants in COVID-19 patients. Clin. Infect. Dis. 2020, 73, e437–e444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 972. [Google Scholar] [CrossRef] [PubMed]
- ATCC. Coronavirus. 2019. Available online: https://www.atcc.org (accessed on 4 February 2022).
- Jia, P.; Dai, S.; Wu, T.; Yang, S. New Approaches to Anticipate the Risk of Reverse Zoonosis. Trends Ecol. Evol. 2021, 36, 580–590. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef]
- Hammer, A.S.; Quaade, M.L.; Rasmussen, T.B.; Fonager, J.; Rasmussen, M.; Mundbjerg, K.; Lohse, L.; Strandbygaard, B.; Jørgensen, C.S.; Alfaro-Núñez, A.; et al. SARS-CoV-2 Transmission between Mink (Neovison vison) and Humans, Denmark. Emerg. Infect. Dis. 2021, 27, 547–551. [Google Scholar] [CrossRef]
- Koopmans, M. SARS-CoV-2 and the human-animal interface: Outbreaks on mink farms. Lancet. Infect. Dis. 2021, 21, 18–19. [Google Scholar] [CrossRef]
- Sit, T.H.C.; Brackman, C.J.; Ip, S.M.; Tam, K.W.S.; Law, P.Y.T.; To, E.M.W.; Yu, V.Y.T.; Sims, L.D.; Tsang, D.N.C.; Chu, D.K.W.; et al. Infection of dogs with SARS-CoV-2. Nature 2020, 586, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Barrs, V.R.; Peiris, M.; Tam, K.W.S.; Law, P.Y.T.; Brackman, C.J.; To, E.M.W.; Yu, V.Y.T.; Chu, D.K.W.; Perera, R.A.P.M.; Sit, T.H.C. SARS-CoV-2 in Quarantined Domestic Cats from COVID-19 Households or Close Contacts, Hong Kong, China. Emerg. Infect. Dis. 2020, 26, 3071–3074. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Elia, G.; Grassi, A.; Giordano, A.; Desario, C.; Medardo, M.; Smith, S.L.; Anderson, E.R.; Prince, T.; Patterson, G.T.; et al. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. Nat. Commun. 2020, 11, 6231. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.-H.; Lee, J.S.-Y.; Saif, L.J.; Gray, G.C. Novel Canine Coronavirus Isolated from a Hospitalized Pneumonia Patient, East Malaysia. Clin. Infect. Dis. 2022, 74, 446–454. [Google Scholar] [CrossRef] [PubMed]
- François, R.; Alexis, D.; Benjamin, R.; Marisa Peyre, V.C. Origin of the COVID-19 Virus: The Trail of Mink Farming. 2021. Available online: https://theconversation.com/origin-of-the-covid-19-virus-the-trail-of-mink-farming-155989 (accessed on 4 February 2022).
- Centers for Disease Control and Prevention. National Center for Emerging and Zoonotic Infectious Diseases (NCEZID). One Health Basics. 2022. Available online: https://www.cdc.gov/ncezid/index.html (accessed on 4 February 2022).
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Salicru, M.; Domingo, E.; Sanchez, A.; Esteban, J.I.; Rodr?guez-Fr?as, F.; Quer, J. Inference with viral quasispecies diversity indices: Clonal and NGS approaches. Bioinformatics 2014, 30, 1104–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, T.; Zhou, W. The third generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 2020, 9, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, S.; Cortese, M.F.; Rodríguez-Algarra, F.; Tabernero, D.; Rando-Segura, A.; Quer, J.; Buti, M.; Rodríguez-Frías, F. Next-generation sequencing for the diagnosis of hepatitis B: Current status and future prospects. Expert Rev. Mol. Diagn. 2021, 21, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Bleidorn, C. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 2016, 14, 1–8. [Google Scholar] [CrossRef]
- Gupta, P.K. Single-molecule DNA sequencing technologies for future genomics research. Trends Biotechnol. 2008, 26, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Keller, M.W.; Rambo-Martin, B.L.; Wilson, M.M.; Ridenour, C.A.; Shepard, S.S.; Stark, T.J.; Neuhaus, E.B.; Dugan, V.G.; Wentworth, D.E.; Barnes, J.R. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Sci. Rep. 2018, 8, 14408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Factor | Examples |
|---|---|
| Degradation of natural and wild spaces | Deforestation. |
| Dam construction. Changes in wildlife migration patterns. Floods due to changes in river boundaries. Global warning, climate change causing the expansion of viral vectors. Agriculture. | |
| Bringing humans closer to wild animals, putative viral reservoirs | Wild animal parks. |
| Hunting. | |
| Tourism/Travel to exotic areas. | |
| Globalization effects | Human mobility losing the quarantine effect. |
| Commercial trade. | |
| Long distance transport of birds and livestock. | |
| Urbanization (concentrating millions of people in small places). | |
| Massive exploitation of animals | Pigs, chicken, birds, livestock, including wild animal farms for fur and food production. |
| Health and social activities | Transfusions, organ transplants. |
| Social changes related to sex and drug abuse. | |
| Large concentrations in closed halls, stadiums, pavilions. |
| Second Generation * | Third Generation ** | |
|---|---|---|
| Read length | 400–≈500 (2 × 300) bp | 60 K–2 M bp |
| Error rates | ≈1%–2.4% | ≈10%–15% |
| Advantages | High throughputs with relatively low error rates makes them suitable for:
| Much longer reads enable more accurate haplotype *** reconstruction, which allows:
|
| Disadvantages | Short reads are poorly suited for:
| Higher error rates difficult characterization of “real” nucleotide substitutions, insertions and deletions. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quer, J.; Colomer-Castell, S.; Campos, C.; Andrés, C.; Piñana, M.; Cortese, M.F.; González-Sánchez, A.; Garcia-Cehic, D.; Ibáñez, M.; Pumarola, T.; et al. Next-Generation Sequencing for Confronting Virus Pandemics. Viruses 2022, 14, 600. https://doi.org/10.3390/v14030600
Quer J, Colomer-Castell S, Campos C, Andrés C, Piñana M, Cortese MF, González-Sánchez A, Garcia-Cehic D, Ibáñez M, Pumarola T, et al. Next-Generation Sequencing for Confronting Virus Pandemics. Viruses. 2022; 14(3):600. https://doi.org/10.3390/v14030600
Chicago/Turabian StyleQuer, Josep, Sergi Colomer-Castell, Carolina Campos, Cristina Andrés, Maria Piñana, Maria Francesca Cortese, Alejandra González-Sánchez, Damir Garcia-Cehic, Marta Ibáñez, Tomàs Pumarola, and et al. 2022. "Next-Generation Sequencing for Confronting Virus Pandemics" Viruses 14, no. 3: 600. https://doi.org/10.3390/v14030600
APA StyleQuer, J., Colomer-Castell, S., Campos, C., Andrés, C., Piñana, M., Cortese, M. F., González-Sánchez, A., Garcia-Cehic, D., Ibáñez, M., Pumarola, T., Rodríguez-Frías, F., Antón, A., & Tabernero, D. (2022). Next-Generation Sequencing for Confronting Virus Pandemics. Viruses, 14(3), 600. https://doi.org/10.3390/v14030600