
Mathematical Modeling Suggests Cooperation of Plant-Infecting Viruses



Abstract
:1. Introduction
2. Materials & Methods
2.1. Data
2.2. Mathematical Models
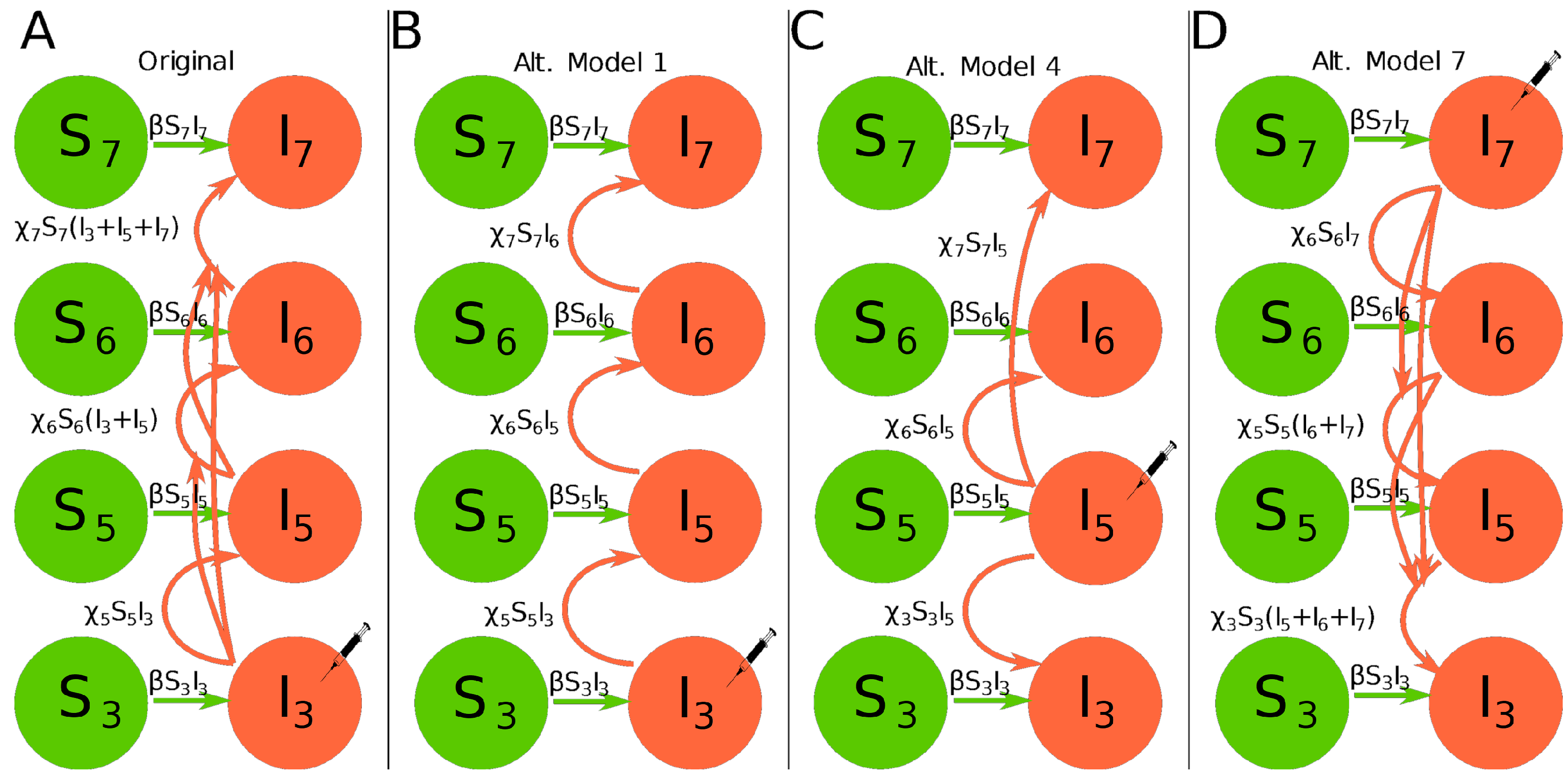
2.2.1. Original Virus Dissemination Model of Tromas et al.
2.2.2. Alternative Virus Dissemination Models for the Total Leaf Infection
- Alternative model 1. In this model the dynamics of infection of the leaf 3 is given in Equation (1), and instead of summing the infection from all the leaves below, we suppose that only the leaf immediately below the one in question can infect it. The dynamics of uninfected leaves is given by Equation (3). Dynamics of infection in other leaves is described by the following equations (see Figure 1B):Initial conditions for the model are if and , otherwise, and . Note that in this model infection of leaf 5 occurs from leaf 3 and not leaf 4.
- Alternative model 2. Leaf 3 infects only leaf 5 which then infects leaves 6 and 7. Leaf 6 also contributes to the infection of leaf 7. Infection for leaf 3 is given by Equation (1) and dynamics of uninfected leaves is given by Equation (3). Dynamics of infection in other leaves is described by the following equations:Initial conditions for the model are if and , otherwise, and . Note that infection of leaf 5 occurs by leaf 3 and not leaf 4.
- Alternative model 3. Infection for leaf 3 is given by Equation (1) and dynamics of uninfected leaves is given by Equation (3). Leaf 3 is the only leave that contributes to infections of higher leaves. Dynamics of infection in other leaves is described by the following equations:Initial conditions for the model are if and , otherwise, and .
- Alternative model 4. The initial infection occurs on leaf 5 which contributes to infections of leaves 3, 6, and 7. All imported virions for these leaves come exclusively from leaf 5. Dynamics of uninfected leaves is given by Equation (3). Dynamics of infection in other leaves is described by the following equations (see Figure 1C):Initial conditions for the model are if and , otherwise, and .
- Alternative model 5. The initial infection occurs on leaf 6 which contributes exclusively to the infections of leaves 3, 5, and 7. Dynamics of uninfected leaves is given by Equation (3) and dynamics of infection in other leaves is described by the following equations:Initial conditions for the model are if and , otherwise, and .
- Alternative model 6. The initial infection occurs on leaf 7 which contributes exclusively to the infections of leaves 3, 5, and 6. Dynamics of uninfected leaves is given by Equation (3) and dynamics of infection in other leaves is described by the following equations:Initial conditions for the model are if and , otherwise, and .
- Alternative model 7. The initial infection occurs on leaf 7 and virus accrues downward; it is essentially the model by Tromas et al. [14] being inverted. Dynamics of uninfected leaves is given by Equation (3) and dynamics of infection in other leaves is described by the following equations (see Figure 1D):Initial conditions for the model are if and , otherwise, and .
- Alternative model 8. The model assumes that infection starts in all leaves and proceeds independently (a.k.a. “logistic” model for individual leaves). Dynamics of infection in all leaves is described by the following equations:Initial conditions are This model has 12 parameters to be estimated from the data.
- Alternative model 9. In all previous models virus dissemination within a given leaf stops when the fraction of infected cells reaches (e.g., Equation (3)). This stop of infection is also observed in the data. However, specific mechanisms of why the infection stops while not all cells in the leaf are infected were not fully investigated. Therefore, in our alternative model we assume that the dynamics of virus infection in a given leaf are not infection level-dependent but instead time-dependent. We define to be the time that the kth leaf accumulates the “immune response” to stop the spread of the virus inside it, and represents how quickly this immune response kicks in [32]. The dynamics of the infection is given by the same equations as in the Tromas et al. [14] model (Equations (1) and (2)), and the dynamics of uninfected cells available for infection due to generation of the immune response in the kth leaf is given bywhere the initial conditions for the model are if and , otherwise. This model has 4 extra parameters as compared to other alternative models but the model can be reduced in size by assuming that some of the parameters (e.g., or ) to be leaf number-independent (see Main text for results). In such cases, the model has 10 parameters to be estimated from the data.
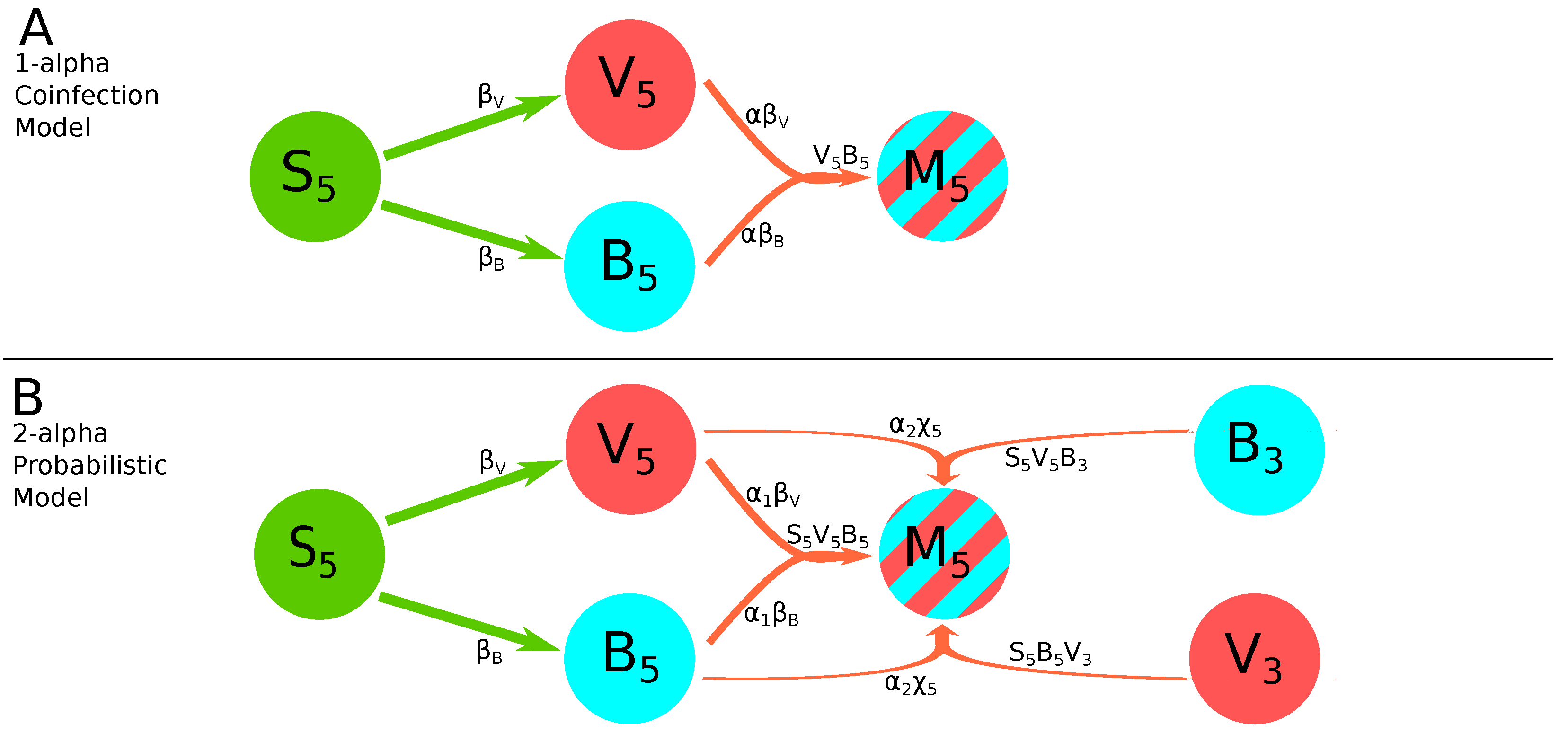
2.2.3. Virus Dissemination Models for the Infection/Coinfection with Two Viral Variants
- 1-alpha coinfection model. In this model, we describe the coinfection growing as dependent on the within-leaf spread dynamics of both viruses. Here, and in other models is proportional to the rate at which coinfections are expected to arise by chance. We sum these these rates assuming that cells are first infected by one variant and then coinfected with another, and use a scaling factor to indicate synergy () or inhibition () of the coinfection process as compared to random, mass action-like infection process:This model has 12 parameters (Figure 2A).
- 2-alpha coinfection model. We assume that the rate of coinfection may proceed differently by the two viral strains denoted by and which is a simple extension of the 1-alpha coinfection model (Equation (21)):This model has 13 parameters.
- Probabilistic model. Because and measure the fraction of cells infected by the particular virus in the kth leaf, then for fraction of coinfected cells, , we can think of the probability of a cell being infected by both strains as being determined by . We can then use parameter to measure how much more or less often coinfection is happening as compared with random chance: means coinfection is behaving like a random process; means coinfection is occurring less often than it would by random chance, and means coinfection is occurring with greater frequency than random chance [33]. Multiplying by the product and differentiating it with respect to t gives:which then with the use of Equations (18) and (19) results in the following model for the dynamics of coinfected cells:This model has 12 parameters. Note that in contrast with previous models (e.g., Equation (22)), in this model coinfection within the leaf depends on the fractions of uninfected () and virus-infected cells in the leaf ( and ).
- 2-alpha probabilistic model. As in the original Tromas et al. [14] model, the equation for coinfection in the probabilistic model is composed of two parts (Equation (24)): the first term with parameters and represents the within-leaf spread, and the second term with the parameter represents the leaf-to-leaf spread. It seemed reasonable that coinfection may be driven more by one form of spread or the other, so we used and to measure their respective contributions:This model has 13 parameters (Figure 2B).
- Logistic model for coinfection growth. The details of how plant cells become coinfected by two different viruses during the local spread are not fully understood. Because typically plant viruses spread to adjacent cells via plasmodesmata, a coinfected cell may be a source of both viral strains when infecting neighboring cells. In this alternative model we therefore assume that the frequency of coinfected cells increases randomly due to viral dissemination systemically from other leaves and logistically due to local, within-leaf spread:This model has 12 parameters.
- 2-alpha logistic model for coinfection growth. Similarly to the 2-alpha probabilistic model, the rate of coinfection may be different between local and systemic viral spread (Equation (25)). Therefore, we use and to differentiate between coinfection occurring as within-leaf and leaf-to-leaf/systemic spread, respectively:This model has 13 parameters.
2.3. Statistical Treatment
2.4. Programming Details
3. Results
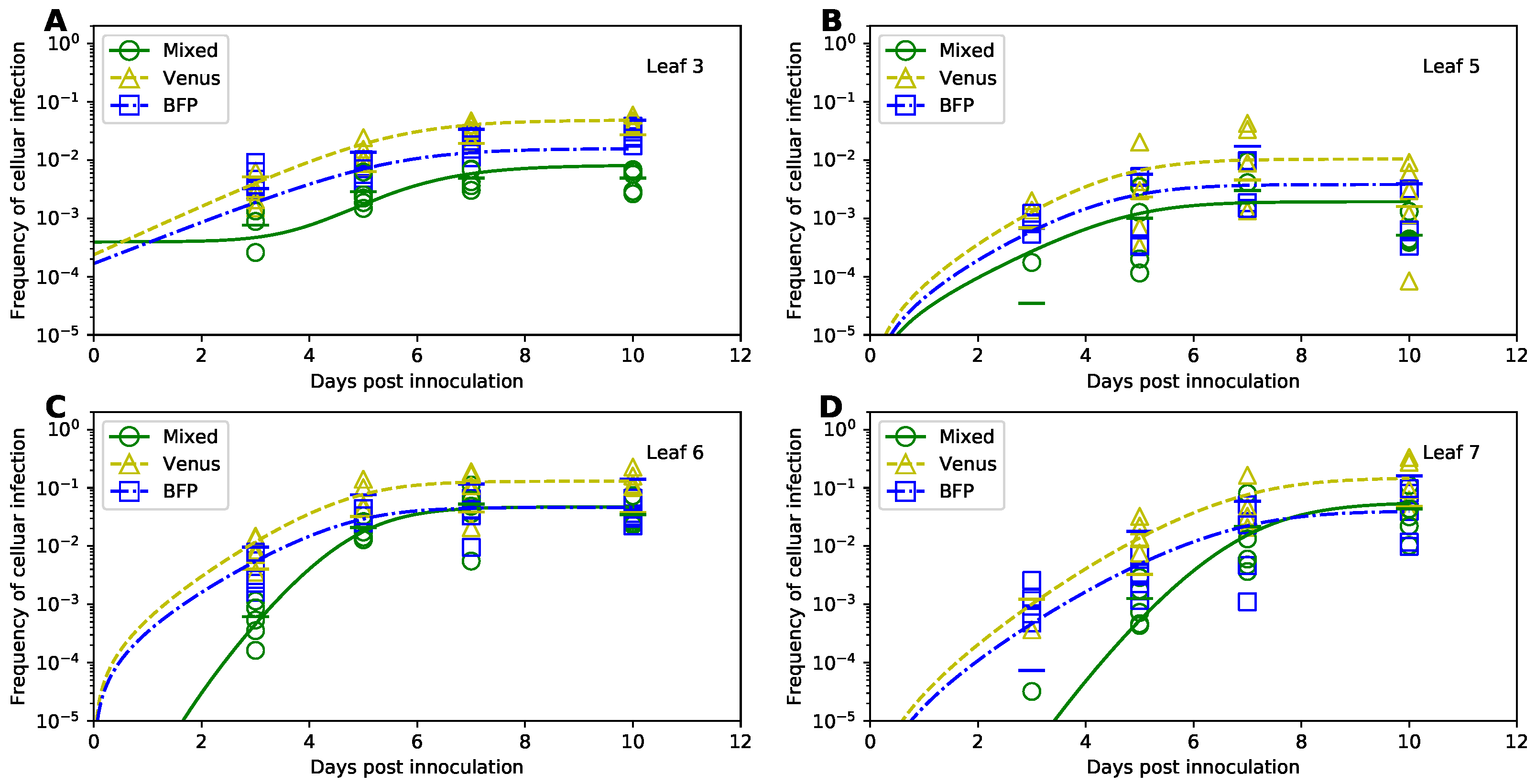
3.1. The Experimental Dataset of the Kinetics of TEV Spread
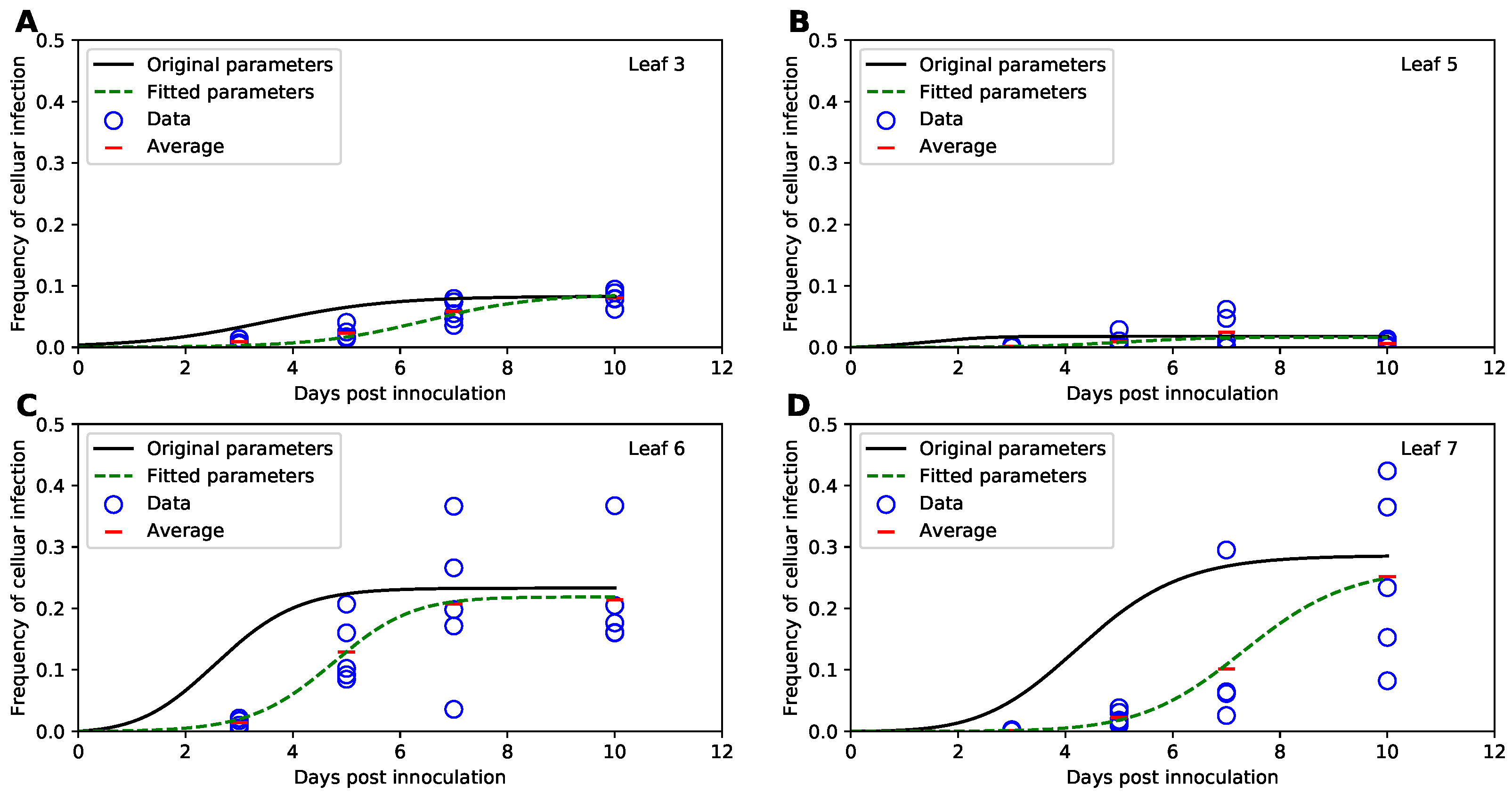
3.2. Model with Tromas et al. Parameter Values Does Not Match the Data
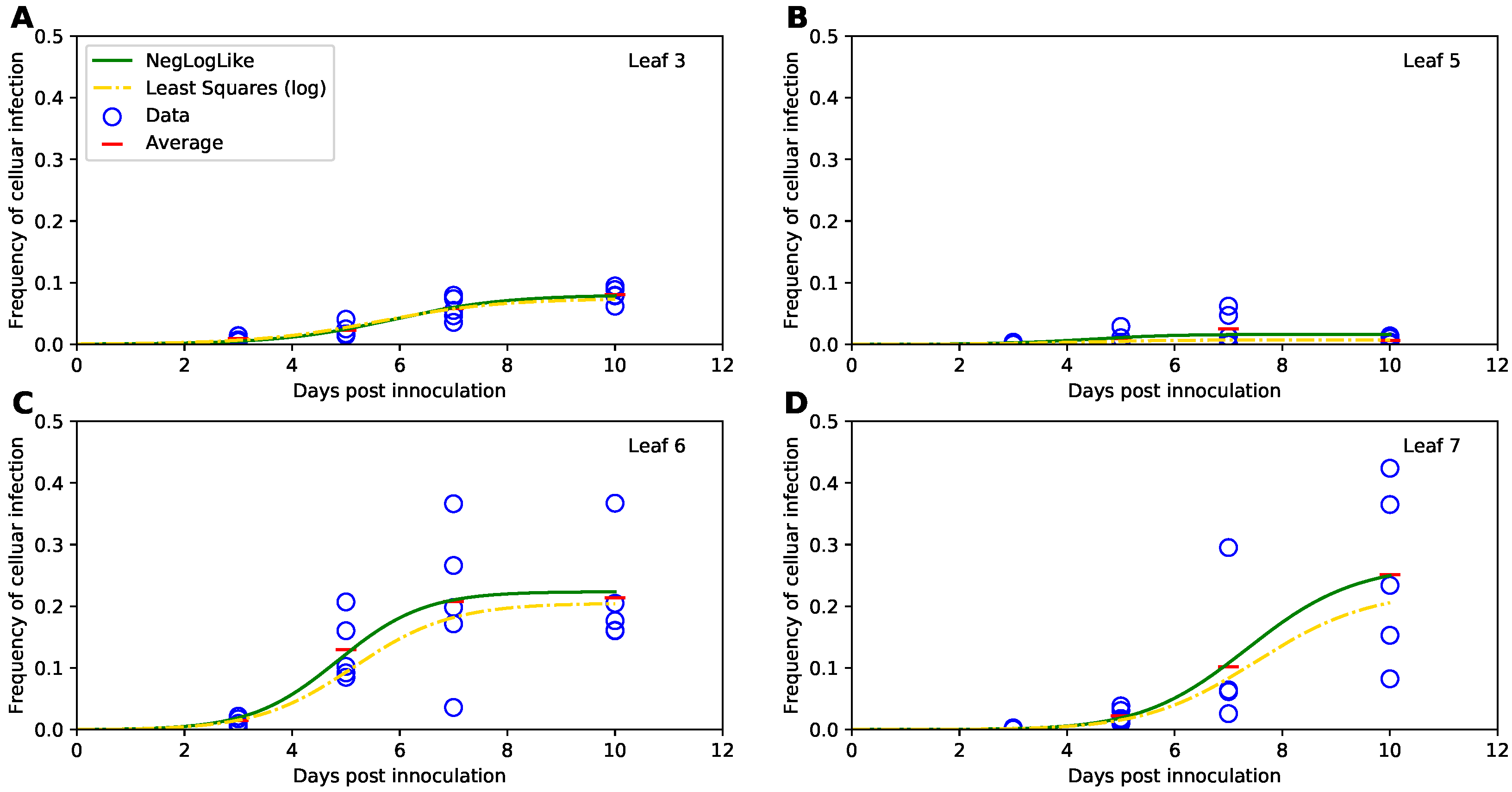
3.3. Fitting the Models Using Binomial Distribution-Based Likelihood or Normal Distribution-Based Likelihood (Least Squares) Delivers Similar Parameter Estimates
3.4. An Alternative Model with Variable Within-Leaf Replication Kinetics Is Consistent with Observed Viral Spread Kinetics
3.5. Alternative Models with Differing Patterns of Viral Dissemination Are Largely Consistent with Observed Viral Spread Kinetics
3.6. Alternative Models Incorporating Independent Replication or Immune Responses Are Also Consistent with Observed Viral Spread Kinetics
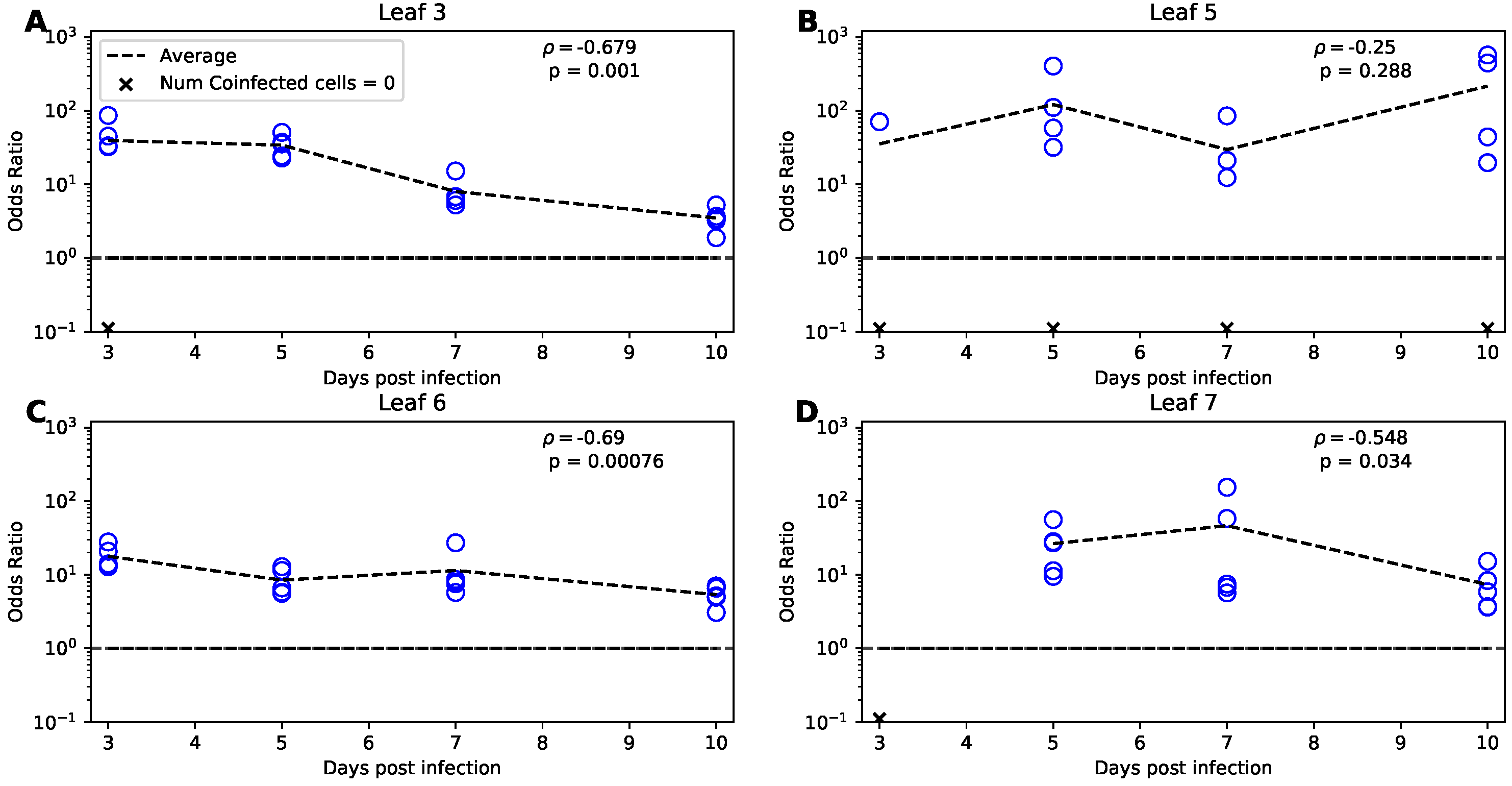
3.7. Odds Ratio Test Implies a Higher Than Random Rate of Coinfection
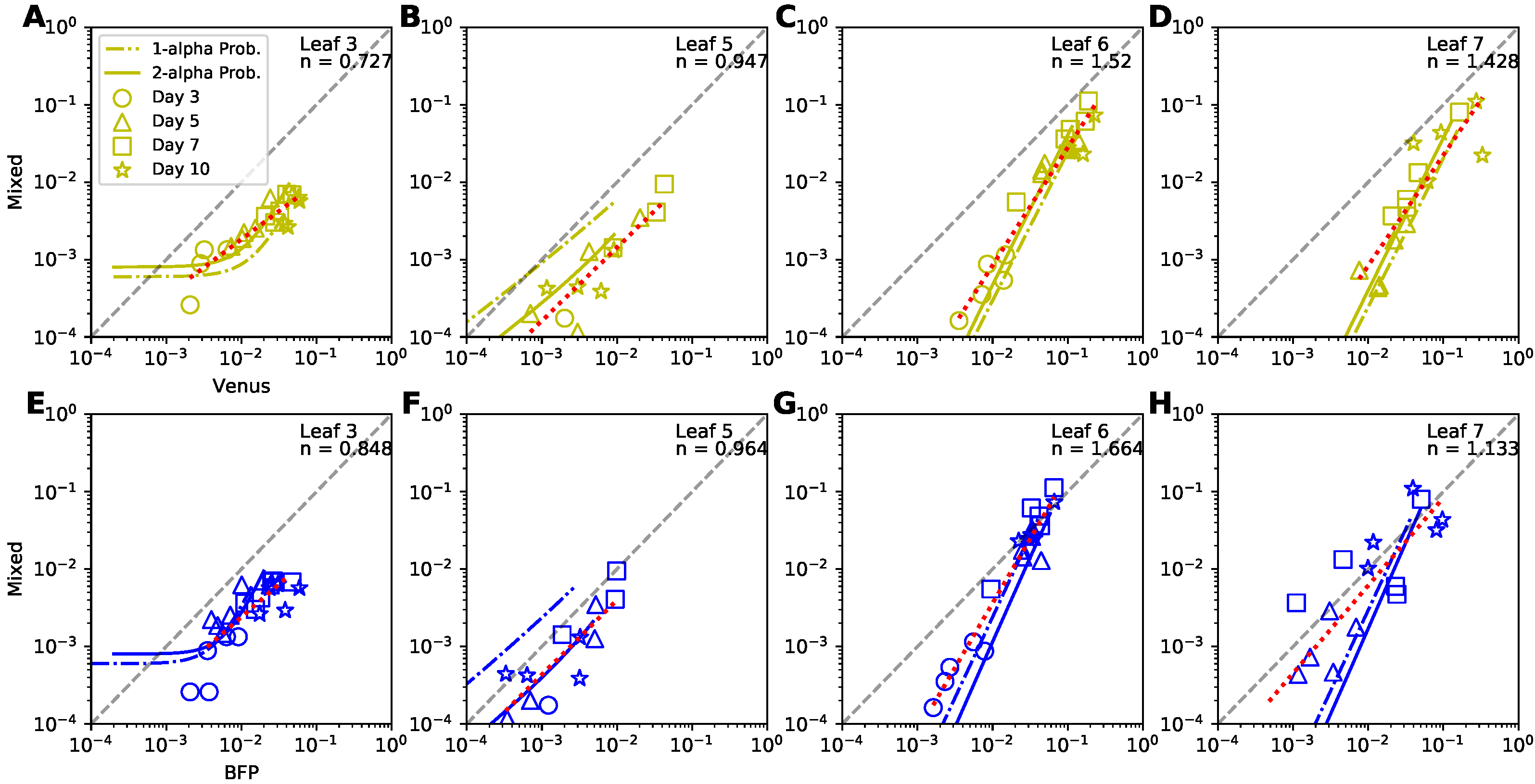
3.8. A Probability-Based Coinfection Model Performs Best Compared to Other Coinfection Models
3.9. Dynamics of Coinfected Cells Compared to Singly-Infected Cells
4. Discussion
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ODE | ordinary differential equations |
| TEV | Tobacco etch virus |
| BFP | blue fluorescent protein |
| negative log-likelihood | |
| LS | least squares |
| LOD | limit of detection |
| AIC | Akaike Information Criterion |
| SSR | sum of squared residuals |
| OR | odds ratio |
References
- Lutz, W.; Sanderson, W.; Scherbov, S. End World Popul. Growth. Nature 2001, 412, 543–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoornweg, D.; Pope, K. Population predictions for the world’s largest cities in the 21st century. Environ. Urban. 2017, 29, 195–216. [Google Scholar] [CrossRef] [Green Version]
- United Nations. Population Facts. 2019. Available online: https://www.un.org/en/development/desa/population/publications/pdf/popfacts/PopFacts_2019-6.pdf (accessed on 14 November 2021).
- Walker, R.J. Population growth and its implications for global security. Am. J. Econ. Sociol. 2016, 4, 980–1004. [Google Scholar] [CrossRef]
- Hull, R. Plant Virology, 5th ed.; Elsevier Science & Technology: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Jones, R.A.C. Global plant virus disease pandemics and epidemics. Plants 2021, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Youden, W.J. Statistical aspect of the production of primary lesions by plant viruses. Nature 1935, 135, 1075. [Google Scholar] [CrossRef]
- Bald, J.G. Statistical aspect of the production of primary lesions by plant viruses. Nature 1935, 135, 996. [Google Scholar] [CrossRef]
- Furumoto, W.A.; Mickey, R. A mathematical model for the infectivity-dilution curve of tobacco mosaic virus: Experimental tests. Virology 1967, 32, 224–233. [Google Scholar] [CrossRef]
- Furumoto, W.A.; Mickey, R. A mathematical model for the infectivity-dilution curve of tobacco mosaic virus: Theoretical considerations. Virology 1967, 32, 216–223. [Google Scholar] [CrossRef]
- Martínez, F.; Sardanyés, J.; Elena, S.F.; Daròs, J.-A. Dynamics of a plant rna virus intracellular accumulation: Stamping machine vs. geometric replication. Genetics 2011, 188, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Lafforgue, G.; Sardanyés, J.; Elena, S.F. Differences in accumulation and virulence determine the outcome of competition during tobacco etch virus coinfection. PLoS ONE 2011, 6, e17917. [Google Scholar] [CrossRef] [Green Version]
- Miljkovic, D.; Depolli, M.; Stare, T.; Mozetič, I.; Petek, M.; Gruden, K.; Lavrač, N. Plant defence model revisions through iterative minimisation of constraint violations. Int. J. Comput. Biol. Drug Des. 2014, 7, 61–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromas, N.; Zwart, M.P.; Lafforgue, G.; Elena, S.F. Within-host spatiotemporal dynamics of plant virus infection at the cellular level. PLoS Genet. 2014, 10, e1004186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.S.; Holt, J.; Colvin, J. Mathematical models of host plant infection by helper-dependent virus complexes: Why are helper viruses always avirulent? Phytopathology 2000, 90, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaku, M.; Burattini, M.N.; Coutinho, F.A.B.; Massad, E. Modeling the competition between viruses in a complex plant-pathogen system. Phytopathology 2010, 100, 1042–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.M.; Manore, C.A.; Bokil, V.A.; Borer, E.T.; Hosseini, P.R. Spatiotemporal model of barley and cereal yellow dwarf virus transmission dynamics with seasonality and plant competition. Bull. Math. Biol. 2011, 70, 2707–2730. [Google Scholar] [CrossRef] [Green Version]
- Jeger, M.J.; Chen, Z.; Powell, G.; Hodge, S.; vanden Bosch, F. Interactions in a host plant-virus-vector-parasitoid system: Modelling the consequences for virus transmission and disease dynamics. Virus Res. 2011, 159, 183–193. [Google Scholar] [CrossRef]
- Neofytou, G.; Kyrychko, Y.N.; Blyuss, K.B. Mathematical model of plant-virus interactions mediated by RNA interference. J. Theor. Biol. 2016, 403, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, F.M.; Allen, L.J.S.; Prendeville, H.R.; Hajimorad, M.R.; Jeger, M.J. The evolution of plant virus transmission pathways. J. Theor. Biol. 2016, 396, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Jeger, M.J.; Madden, L.V.; vanden Bosch, F. Plant virus epidemiology: Applications and prospects for mathematical modeling and analysis to improve understanding and disease control. Plant Dis. 2018, 102, 837–854. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.H.; Gómez-Gardeñes, J.; Meloni, S.; Estrada, E. Epidemics on plants: Modeling long-range dispersal on spatially embedded networks. J. Theor. Biol. 2018, 453, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Allen, L.J.S.; Bokil, V.A.; Cunniffe, N.J.; Hamelin, F.M.; Hilker, F.M.; Jeger, M.J. Modelling vector transmission and epidemiology of co-infecting plant viruses. Viruses 2019, 11, 1153. [Google Scholar] [CrossRef] [Green Version]
- Kendig, A.E.; Borer, E.T.; Boak, E.N.; Picard, T.C.; Seabloom, E.W. Host nutrition mediates interactions between plant viruses, altering transmission and predicted disease spread. Ecology 2020, 101, e03155. [Google Scholar] [CrossRef] [PubMed]
- AlBasir, F.; Kyrychko, Y.N.; Blyuss, K.B.; Ray, S. Effects of vector maturation time on the dynamics of cassava mosaic disease. Bull. Math. Biol. 2021, 83, 87. [Google Scholar] [CrossRef] [PubMed]
- Reagan, B.C.; Burch-Smith, T.M. Viruses reveal the secrets of plasmodesmal cell biology. Mol. Plant-Microbe Interact. 2020, 33, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, R.; LaCamera, S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.-L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L.; et al. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.G.; Cruz, S.S.; Roberts, I.M.; Prior, D.A.M.; Turgeon, R.; Oparka, K.J. Phloem unloading in sink leaves of nicotiana benthamiana: Comparison of a fluorescent solute with a fluorescent virus. Plant Cell 1997, 9, 1381–1396. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hajimorad, M.R. Gain of virulence by soybean mosaic virus on rsv4-genotype soybeans is associated with a relative fitness loss in a susceptible host. Mol. Plant Pathol. 2016, 17, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Burnham, K.P.; Anderson, D.R. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach; Springer: New York, NY, USA, 2002. [Google Scholar]
- Creager, A.N. The Life of a Virus: Tobacco Mosaic Virus as an Experimental Model, 1930–1965; University of Chicago Press: Chicago, IL, USA, 2002. [Google Scholar]
- Gupta, N.; Reddy, K.; Bhattacharyya, D.; Chakraborty, S. Plant responses to geminivirus infection: Guardians of the plant immunity. Virol. J. 2021, 18, 143. [Google Scholar] [CrossRef]
- Dang, Q.; Chen, J.; Unutmaz, D.; Coffin, J.M.; Pathak, V.K.; Powell, D.; KewalRamani, V.N.; Maldarelli, F.; Hu, W.-S. Nonrandom HIV-1 infection and double infection via direct and cell-mediated pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Efron, B.; Tibshirani, R. An Introduction to the Bootstrap; Chapman & Hall: New York, NY, USA, 1993. [Google Scholar]
- Wang, A. Cell-to-cell movement of plant viruses via plasmodesmata: A current perspective on potyviruses. Curr. Opin. Virol. 2021, 48, 10–16. [Google Scholar] [CrossRef]
- Wu, F.-H.; Shen, S.-C.; Lee, L.-Y.; Lee, S.-H.; Chan, M.-T.; Lin, C.-S. Tape-Arabidopsis sandwich—A simpler Arabidopsis protoplast isolation method. Plant Methods 2009, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganusov, V.V. Strong Inference in Mathematical Modeling: A Method for Robust Science in the Twenty-First Century. Front. Microbiol. 2016, 7, 1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffin, J.M. HIV population dynamics invivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Letvin, N.L.; Walker, B.D. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat. Med. 2003, 9, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Giorgi, E.E.; Ganusov, V.V.; Cai, F.; Athreya, G.; Yoon, H.; Carja, O.; Hora, B.; Hraber, P.; Romero-Severson, E.; et al. Tracking HIV-1 recombination to resolve its contribution to HIV-1 evolution in natural infection. Nat. Commun. 2018, 9, 1928. [Google Scholar] [CrossRef] [Green Version]
- Haqqani, A.A.; Marek, S.L.; Kumar, J.; Davenport, M.; Wang, H.; Tilton, J.C. Central memory CD4+ T cells are preferential targets of double infection by HIV-1. Virol. J. 2015, 12, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-B.; Metzlaff, M. RNA silencing and antiviral defense in plants. Curr. Opin. Plant Biol. 2005, 8, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Anandalakshmi, R.; Pruss, G.J.; Ge, X.; Marathe, R.; Mallory, A.C.; Smith, T.H.; Vance, V.B. A viral suppressor of gene silencing in plants. Proc. Natl. Acad. Sci. USA 1998, 95, 13079–13084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csorba, T.; Kontra, L.; Burgyán, J. viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology 2015, 479–480, 85–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, S.; Pirolles, E.; Yvon, M.; Baecker, V.; Michalakis, Y.; Blanc, S. The multiplicity of cellular infection changes depending on the route of cell infection in a plant virus. J. Virol. 2015, 89, 9665–9675. [Google Scholar] [CrossRef] [Green Version]
- Dixit, N.; Perelson, A. HIV dynamics with multiple infections of target cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8198–8203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavaliev, R.; Ueki, S.; Epel, B.L.; Citovsky, V. Biology of callose (beta-1,3-glucan) turnover at plasmodesmata. Protoplasma 2011, 248, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.R.; Wilson, A.K. Transcomplementation and synergism in plants: Implications for viral transgenes? Mol. Plant Pathol. 2008, 9, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Fondong, V.N. The search for resistance to cassava mosaic geminiviruses: How much we have accomplished, and what lies ahead. Front. Plant Sci. 2017, 8, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, S.; Yvon, M.; Thébaud, G.; Monsion, B.; Michalakis, Y.; Blanc, S. Dynamics of the multiplicity of cellular infection in a plant virus. PLoS Pathog. 2010, 6, e1001113. [Google Scholar] [CrossRef] [Green Version]
- Andreu-Moreno, I.; Bou, J.-V.; Sanjuán, R. Cooperative nature of viral replication. Sci. Adv. 2020, 6, eabd4942. [Google Scholar] [CrossRef]
- Díaz-Munoz, S.L.; Sanjuán, R.; West, S. Sociovirology: Conflict, cooperation, and communication among viruses. Cell Host Microbe 2017, 22, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R. The social life of viruses. Annu. Rev. Virol. 2021, 8, 183–199. [Google Scholar] [CrossRef]
- Graw, F.; Balagopal, A.; Kandathil, A.J.; Ray, S.C.; Thomas, D.L.; Ribeiro, R.M.; Perelson, A.S. Inferring viral dynamics in chronically HCV infected patients from the spatial distribution of infected hepatocytes. PLoS Comput. Biol. 2014, 10, e1003934. [Google Scholar] [CrossRef] [Green Version]
- Graw, F.; Perelson, A.S. Modeling viral spread. Annu. Rev. Virol. 2016, 3, 555–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durso-Cain, K.; Kumberger, P.; Schälte, Y.; Fink, T.; Dahari, H.; Hasenauer, J.; Uprichard, S.L.; Graw, F. HCV spread kinetics reveal varying contributions of transmission modes to infection dynamics. Viruses 2021, 13, 1308. [Google Scholar] [CrossRef]
- Gallagher, M.E.; Brooke, C.B.; Ke, R.; Koelle, K. Causes and consequences of spatial within-host viral spread. Viruses 2018, 10, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirouette, C.; Younis, N.P.; Reddy, M.B.; Beauchemin, C.A.A. A mathematical model describing the localization and spread of influenza a virus infection within the human respiratory tract. PLoS Comput. Biol. 2020, 16, e1007705. [Google Scholar] [CrossRef]
- Stiefel, P.; Schmidt, F.I.; Dörig, P.; Behr, P.; Zambelli, T.; Vorholt, J.A.; Mercer, J. Cooperative vaccinia infection demonstrated at the single-cell level using fluidfm. Nano Lett. 2012, 12, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Tauzin, A.; Remion, A.; Ejima, K.; Mammano, F.; Iwami, S. Dynamics of HIV-1 coinfection in different susceptible target cell populations during cell-free infection. J. Theor. Biol. 2018, 455, 39–46. [Google Scholar] [CrossRef]
- Deleage, C.; Immonen, T.T.; Fennessey, C.M.; Reynaldi, A.; Reid, C.; Newman, L.; Lipkey, L.; Schlub, T.E.; Camus, C.; O’Brien, S.; et al. Defining early SIV replication and dissemination dynamics following vaginal transmission. Sci. Adv. 2019, 5, eaav7116. [Google Scholar] [CrossRef] [Green Version]
- Kappagantu, M.; Collum, T.D.; Dardick, C.; Culver, J.N. Viral hacks of the plant vasculature: The role of phloem alterations in systemic virus infection. Annu. Rev. Virol. 2020, 7, 351–370. [Google Scholar] [CrossRef]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyrt, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Gern, B.H.; Adams, K.N.; Plumlee, C.R.; Stoltzfus, C.R.; Shehata, L.; Moguche, A.O.; Busman-Sahay, K.; Hansen, S.G.; Axthelm, M.K.; Picker, L.J.; et al. TGFβ restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe 2021, 29, 594–606.e6. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Wang, A. Research advances in Potyviruses: From the laboratory bench to the field. Annu. Rev. Phytopathol. 2021, 59, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Wisler, G.C.; Duffus, J.E. A century of plant virus management in the Salinas valley of California, ‘East of Eden’. Virus Res. 2000, 71, 161–169. [Google Scholar] [CrossRef]
- Khatabi, B.; Fajolu, O.L.; Wen, R.-H.; Hajimorad, M.R. Evaluation of North American isolates of Soybean mosaic virus for gain of virulence on Rsv-genotype soybeans with special emphasis on resistance-breaking determinants on Rsv4. Mol. Plant Pathol. 2012, 13, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Ganusova, E.E.; Burch-Smith, T.M. Review: Plant-pathogen interactions through the plasmodesma prism. Plant Sci. 2019, 279, 70–80. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Parameters () | New Parameters () | New Parameters () | ||||
|---|---|---|---|---|---|---|
| Parameter | Estimate | 95% CIs | Estimate | 95% CIs | Estimate | 95% CIs |
| 0.00372 | (0.001,0.017) | 0.00022 | (0.00011,0.00108) | 0.0005 | (0.0001,0.0008) | |
| , 1/day | 0.871 | (0.257,1.66) | 0.950 | (0.549,1.183) | 0.902 | (0.730,1.618) |
| , 1/day | 0.724 | (0.033,0.813) | 0.167 | (0.042,17.291) | 0.063 | (0.023,0.162) |
| , 1/day | 1.38 | (0.580,2.340) | 1.046 | (0.228,1.487) | 0.691 | (0.387,1.053) |
| , 1/day | 0.107 | (0.050,0.263) | 0.029 | (0.009,0.160) | 0.029 | (0.015,0.051) |
| 0.083 | (0.053,0.147) | 0.080 | (0.074,0.134) | 0.074 | (0.051,0.096) | |
| 0.018 | (0.002,0.050) | 0.016 | (0.005,0.024) | 0.006 | (0.004,0.010) | |
| 0.233 | (0.155,0.345) | 0.224 | (0.203,0.287) | 0.204 | (0.181,0.234) | |
| 0.286 | (0.234,0.346) | 0.269 | (0.130,0.418) | 0.224 | (0.092,0.557) | |
| Fitted with Least Squares Method (Log Transformation & 0 ≡ LOD) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Original | Alt. | Alt. | Alt. | Alt. | Alt. | Alt. | Alt. |
| Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 | Model 7 | ||
| 0.0005 | 0.0008 | 0.0006 | 0.0001 | 0.00006 | 0.0008 | 0.0005 | 0.0005 | |
| , 1/day | 0.902 | 0.744 | 0.871 | 1.299 | 1.050 | 1.028 | 1.116 | 1.159 |
| , 1/day | N/A | N/A | N/A | N/A | 1.672 | 0.133 | 1.541 | 0.286 |
| , 1/day | 0.063 | 0.059 | 0.059 | 0.322 | N/A | 0.025 | 0.277 | 0.050 |
| , 1/day | 0.691 | 8.201 | 8.025 | 3.987 | 3.663 | N/A | 3.079 | 3.263 |
| , 1/day | 0.029 | 0.073 | 0.749 | 0.477 | 0.332 | 0.026 | N/A | N/A |
| 0.074 | 0.083 | 0.075 | 0.072 | 0.060 | 0.059 | 0.055 | 0.051 | |
| 0.006 | 0.006 | 0.006 | 0.005 | 0.006 | 0.006 | 0.005 | 0.005 | |
| 0.204 | 0.204 | 0.199 | 0.194 | 0.201 | 0.215 | 0.189 | 0.184 | |
| 0.224 | 0.228 | 0.238 | 13.000 | 0.215 | 0.211 | 0.218 | 0.209 | |
| 52.713 | 51.991 | 52.255 | 55.612 | 54.468 | 55.148 | 55.486 | 56.309 | |
| −15 | −16 | −16 | −11 | −13 | −12 | −11 | −10 | |
| 1 | 0 | 0 | 5 | 3 | 4 | 5 | 6 | |
| W | 0.963 | 0.959 | 0.960 | 0.973 | 0.969 | 0.973 | 0.972 | 0.973 |
| p | 0.021 | 0.012 | 0.013 | 0.099 | 0.049 | 0.091 | 0.080 | 0.084 |
| Parameters | 1- Probabilistic | 2- Probabilistic | 1- Coinfection | 2- Coinfection | 1- Logistic | 2- Logistic |
|---|---|---|---|---|---|---|
| Equation (24) | Equation (25) | Equation (21) | Equation (22) | Equation (26) | Equation (27) | |
| 512,112 | 511,728 | 519,132 | 525,576 | 515,892 | 517,656 | |
| 1,024,246 | 1,023,480 | 1,038,286 | 1,051,176 | 1,031,806 | 1,035,336 | |
| 766 | 0 | 14,806 | 27,696 | 8326 | 11,856 | |
| 339.459 | 269.521 | 335.169 | 514.719 | 421.662 | 477.806 | |
| 109 | 56 | 106 | 211 | 161 | 193 | |
| 53 | 0 | 50 | 155 | 105 | 137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, J.; Burch-Smith, T.M.; Ganusov, V.V. Mathematical Modeling Suggests Cooperation of Plant-Infecting Viruses. Viruses 2022, 14, 741. https://doi.org/10.3390/v14040741
Miller J, Burch-Smith TM, Ganusov VV. Mathematical Modeling Suggests Cooperation of Plant-Infecting Viruses. Viruses. 2022; 14(4):741. https://doi.org/10.3390/v14040741
Chicago/Turabian StyleMiller, Joshua, Tessa M. Burch-Smith, and Vitaly V. Ganusov. 2022. "Mathematical Modeling Suggests Cooperation of Plant-Infecting Viruses" Viruses 14, no. 4: 741. https://doi.org/10.3390/v14040741
APA StyleMiller, J., Burch-Smith, T. M., & Ganusov, V. V. (2022). Mathematical Modeling Suggests Cooperation of Plant-Infecting Viruses. Viruses, 14(4), 741. https://doi.org/10.3390/v14040741