
EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Candidate EGR1 Target Genes
2.2. Cell Culture
2.3. Viruses and Viral Infections
2.4. Western Blot Analysis
2.5. Transfections
2.6. Generation of EGR1−/− U87MG Cells
2.7. RNA Isolation and RT-qPCR
2.8. Plaque Assays
2.9. Drug Treatments and CellTiter-Glo Assays
2.10. Statistics
3. Results
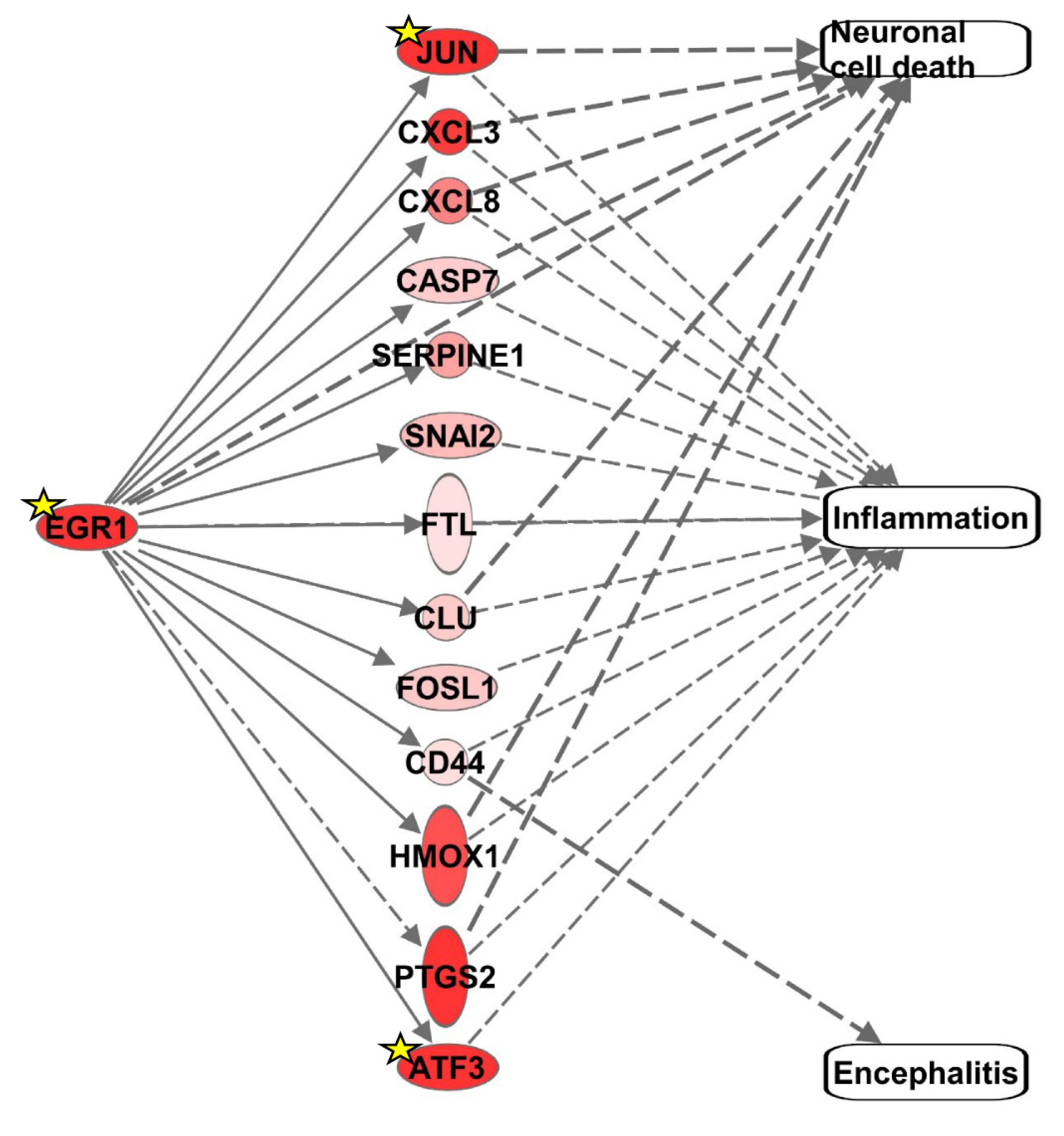
3.1. Identification of Differentially Expressed Genes (DEGs) Associated with Neuronal Cell Death, Inflammation, or Encephalitis and Consistent with EGR1 Upregulation in VEEV-Infected Cells
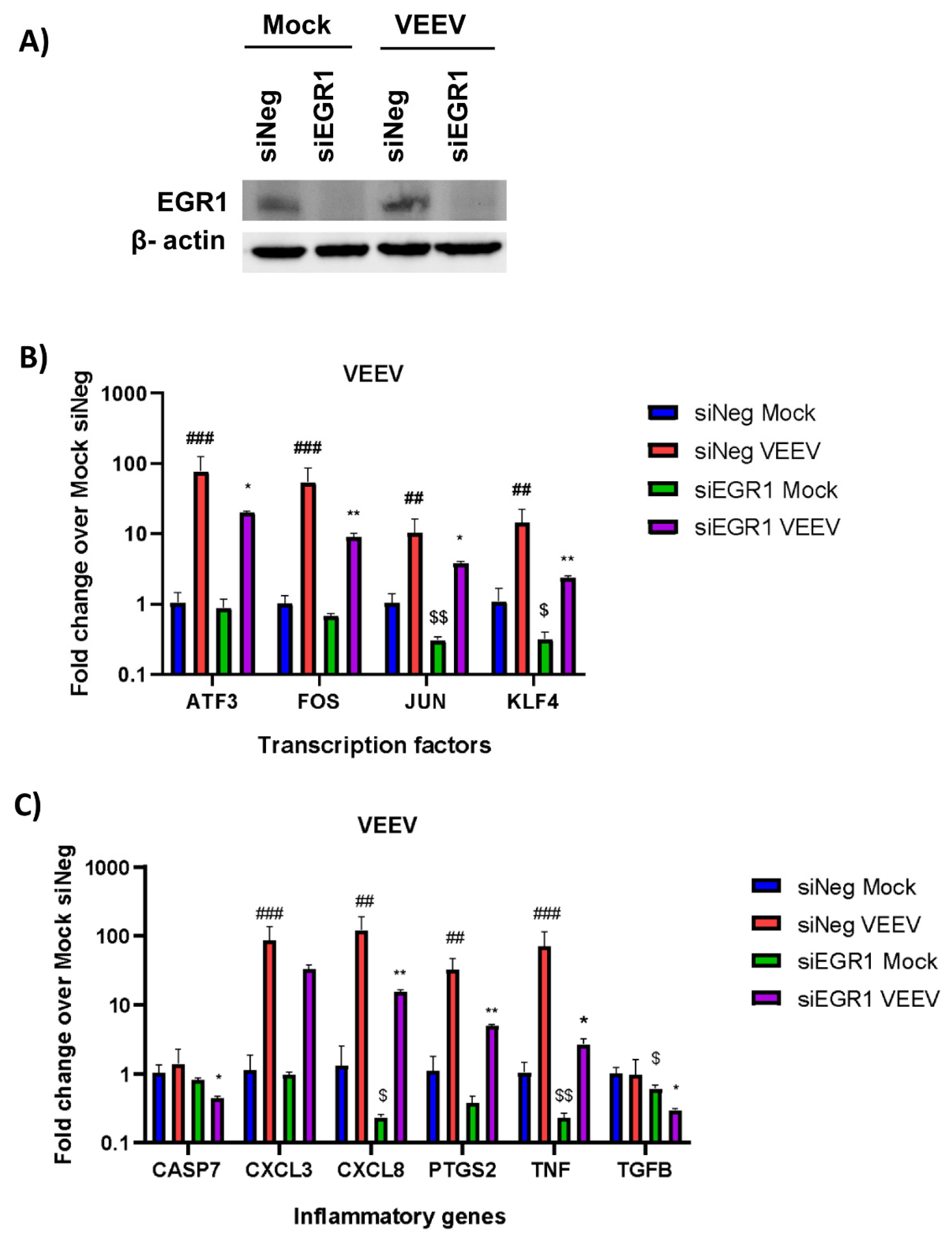
3.2. Gene Expression of Multiple Inflammatory Mediators and Transcription Factors Are Dependent on EGR1 following VEEV Infection
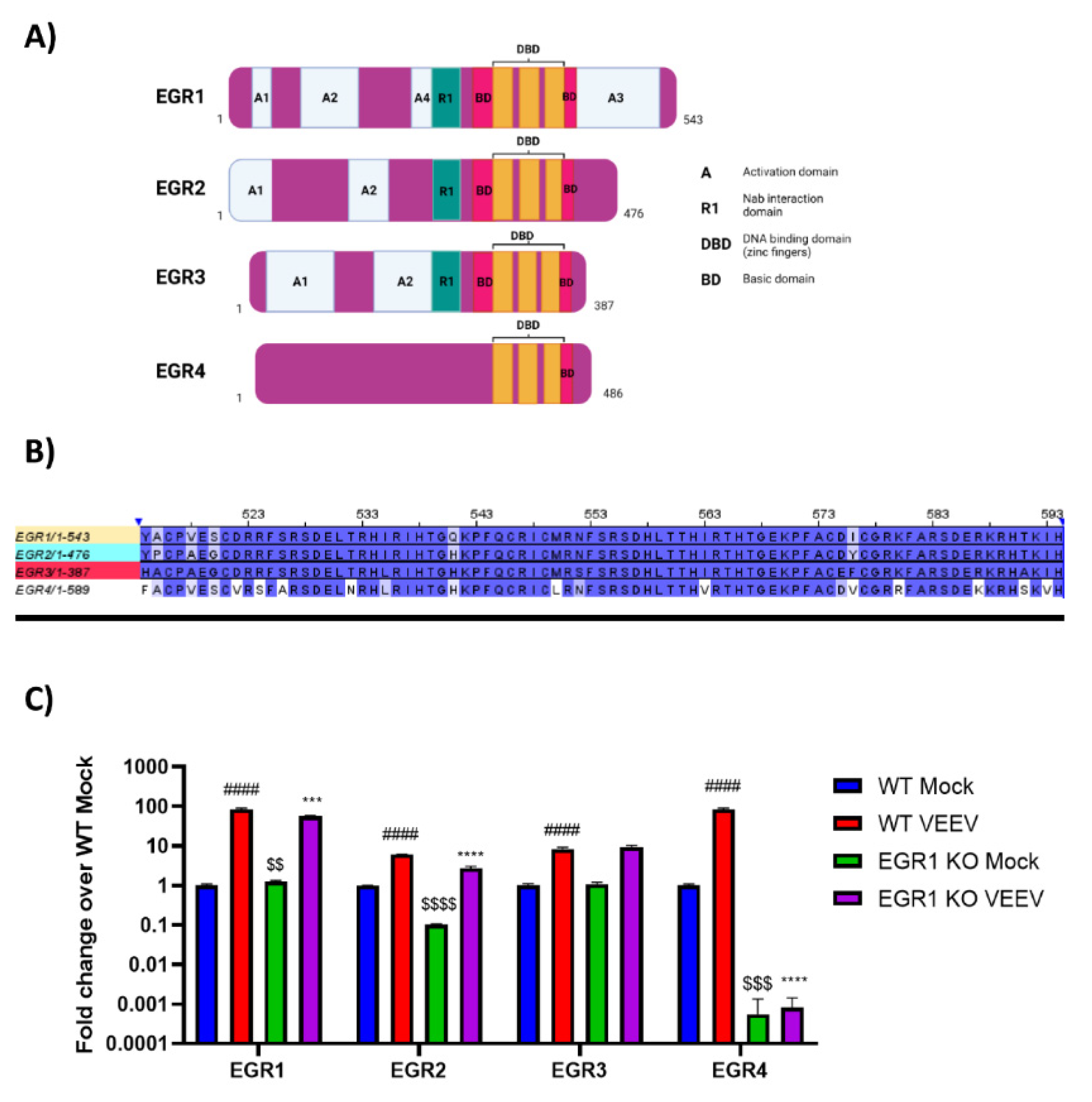
3.3. EGR Family Members Are Regulated by EGR1
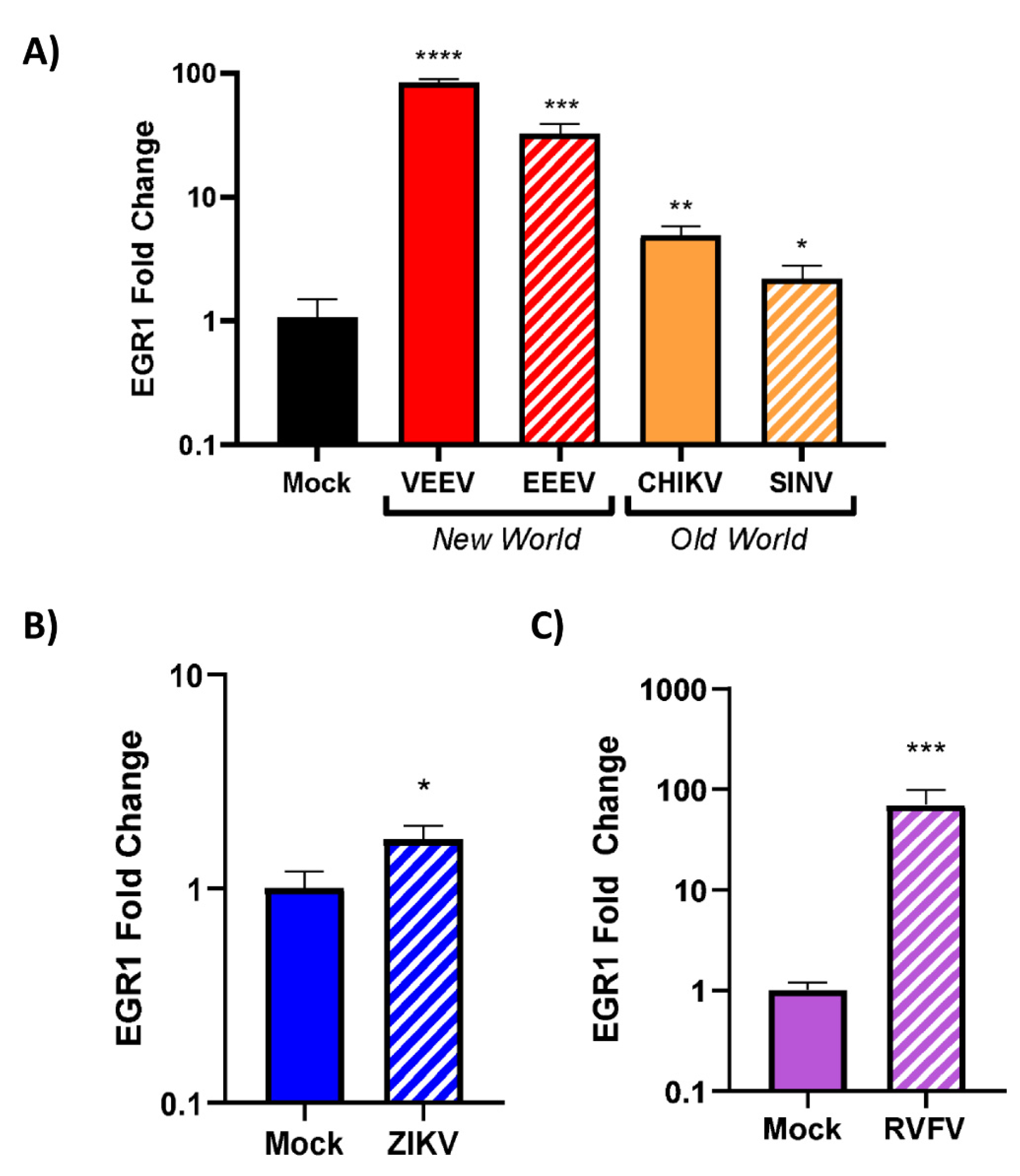
3.4. EGR1 Is Upregulated in VEEV-, EEEV-, SINV-, CHIKV-, ZIKV-, and RVFV-Infected Cells
3.5. Loss of EGR1 Has Minimal Impact on VEEV, EEEV, CHIKV, SINV, RVFV, and ZIKV Viral Titers
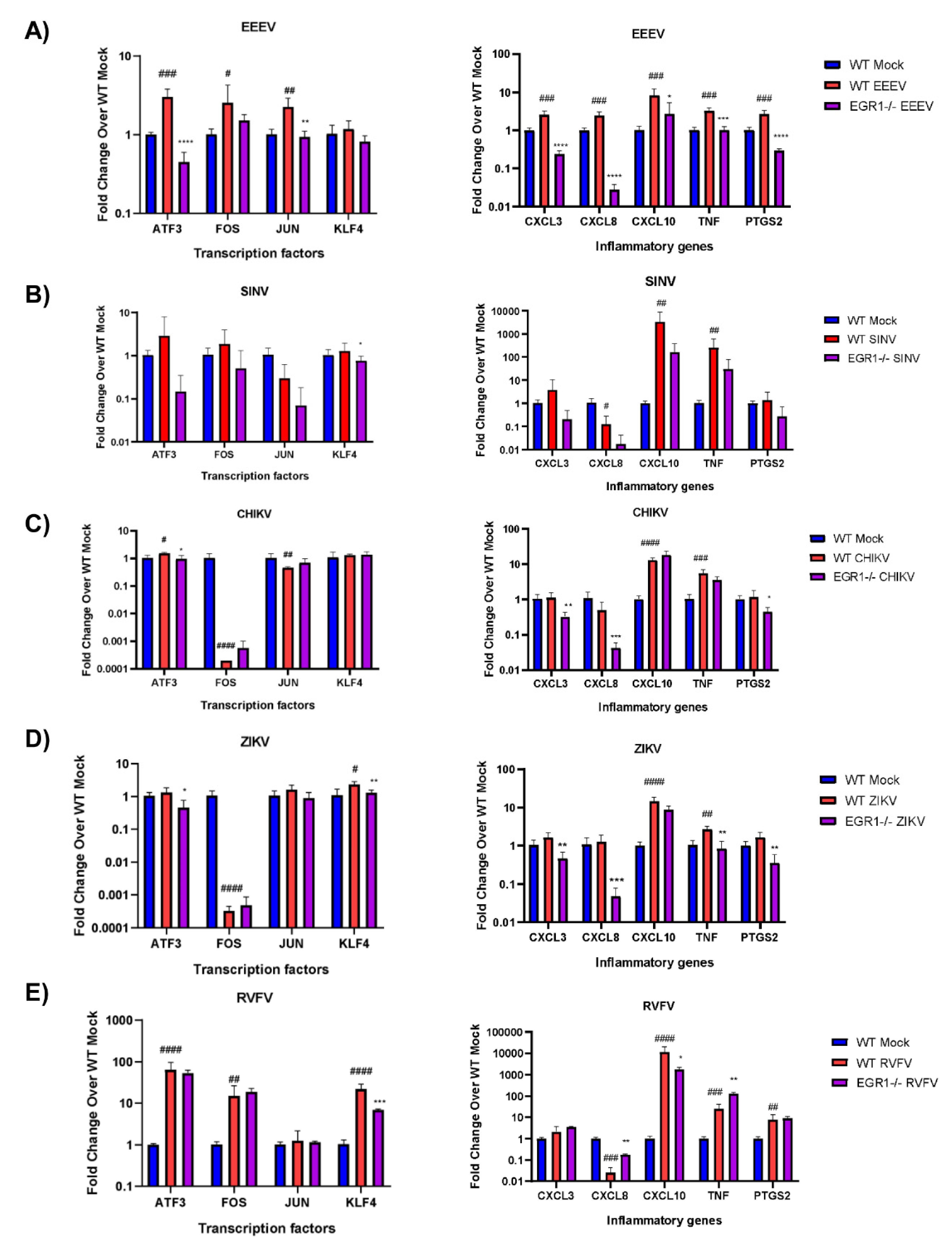
3.6. EGR1-Dependent Gene Expression in EEEV-, SINV-, CHIKV-, ZIKV-, and RVFV-Infected Cells
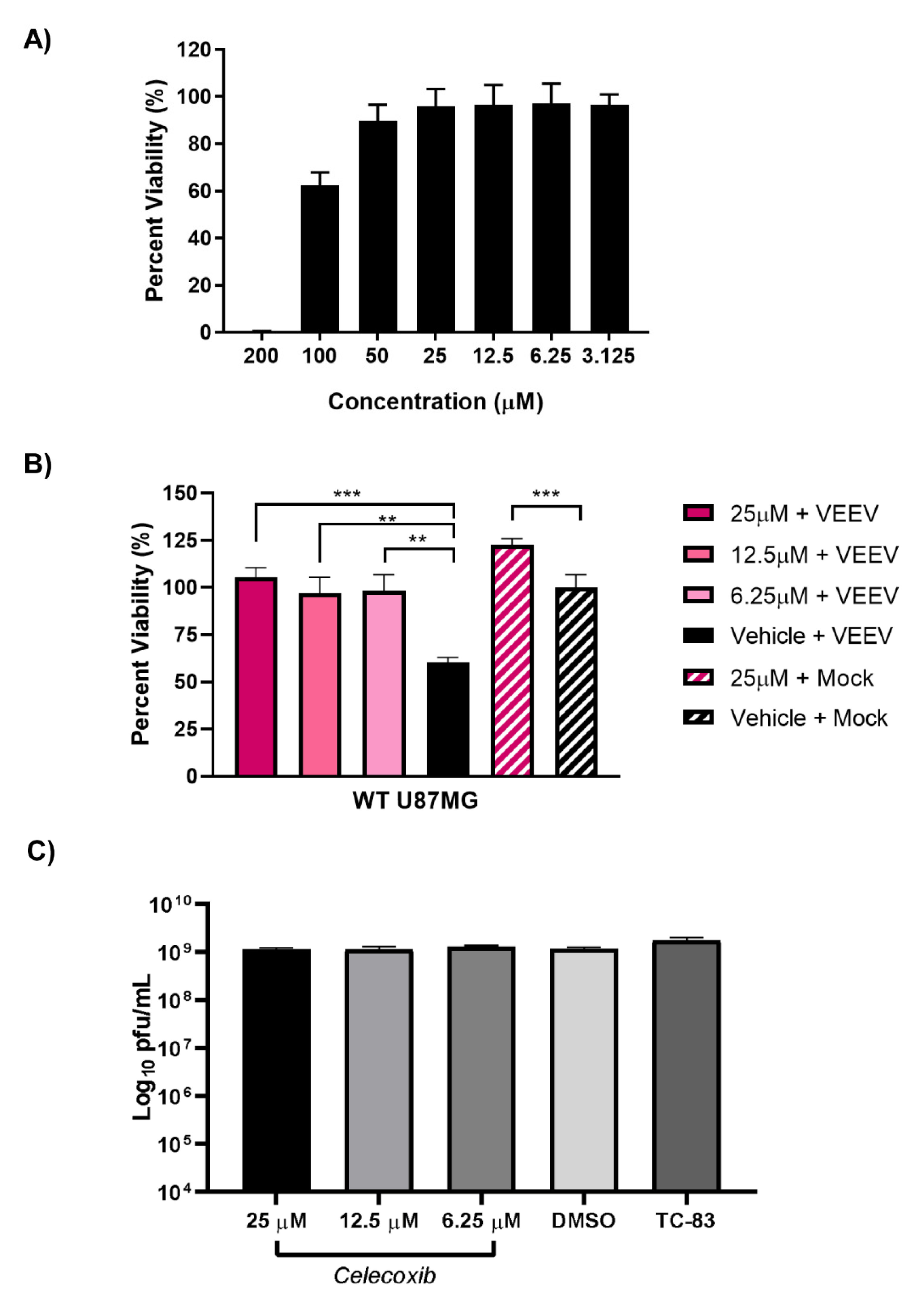
3.7. Inhibition of PTGS2 with Celecoxib Rescues Cells from VEEV-Induced Cells Death but has No Effect on Viral Titers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryman, K.D.; Klimstra, W.B. Host responses to alphavirus infection. Immunol. Rev. 2008, 225, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.; Weaver, S.; Watowich, S.; Chiu, W. Structural biology of old world and new world alphaviruses. Arch. Virol. Suppl. 2005, 19, 179–185. [Google Scholar]
- Azar, S.R.; Campos, R.K.; Bergren, N.A.; Camargos, V.N.; Rossi, S.L. Epidemic Alphaviruses: Ecology, Emergence and Outbreaks. Microorganisms 2020, 8, 1167. [Google Scholar] [CrossRef] [PubMed]
- Ronca, S.E.; Dineley, K.T.; Paessler, S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2016, 7, 959. [Google Scholar] [CrossRef]
- Baer, A.; Lundberg, L.; Swales, D.; Waybright, N.; Pinkham, C.; Dinman, J.D.; Jacobs, J.L.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Induces Apoptosis through the Unfolded Protein Response Activation of EGR1. J. Virol. 2016, 90, 3558–3572. [Google Scholar] [CrossRef] [Green Version]
- Duclot, F.; Kabbaj, M. The Role of Early Growth Response 1 (EGR1) in Brain Plasticity and Neuropsychiatric Disorders. Front. Behav. Neurosci. 2017, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Thiel, G.; Mayer, S.I.; Müller, I.; Stefano, L.; Rössler, O.G. Egr-1-A Ca(2+)-regulated transcription factor. Cell Calcium 2010, 47, 397–403. [Google Scholar] [CrossRef]
- Ho, L.-C.; Sung, J.-M.; Shen, Y.-T.; Jheng, H.-F.; Chen, S.-H.; Tsai, P.-J.; Tsai, Y.-S. Egr-1 deficiency protects from renal inflammation and fibrosis. J. Mol. Med. 2016, 94, 933–942. [Google Scholar] [CrossRef]
- Yan, S.F.; Fujita, T.; Lu, J.; Okada, K.; Shan Zou, Y.; Mackman, N.; Pinsky, D.J.; Stern, D.M. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat. Med. 2000, 6, 1355–1361. [Google Scholar] [CrossRef]
- Harja, E.; Bucciarelli, L.G.; Lu, Y.; Stern, D.M.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Early growth response-1 promotes atherogenesis: Mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ. Res. 2004, 94, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Martín, D.; Díaz-Zamudio, M.; Galindo-Campos, M.; Alcocer-Varela, J. Early growth response transcription factors and the modulation of immune response: Implications towards autoimmunity. Autoimmun. Rev. 2010, 9, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M. Early Growth Response-1, an Integrative Sensor in Cardiovascular and Inflammatory Disease. J. Am. Heart Assoc. 2021, 10, e023539. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Fan, Y.; de Belle, I.; Niemeyer, C.; Gottardis, M.M.; Mercola, D.; Adamson, E.D. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int. J. Cancer 1997, 72, 102–109. [Google Scholar] [CrossRef]
- Virolle, T.; Adamson, E.D.; Baron, V.; Birle, D.; Mercola, D.; Mustelin, T.; de Belle, I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 2001, 3, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Lin, P.C.; Chen, S.P.; Lin, C.C.; Tsai, N.M.; Cheng, Y.L.; Chang, W.L.; Lin, S.Z.; Harn, H.J. Activation of nonsteroidal anti-inflammatory drug-activated gene-1 via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase revealed a isochaihulactone-triggered apoptotic pathway in human lung cancer A549 cells. J. Pharmacol. Exp. Ther. 2007, 323, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.Y.; Kim, G.Y.; Kim, N.D.; Jung, J.H.; Kim, S.K.; Kang, H.S.; Choi, Y.H. Induction of apoptosis by pectenotoxin-2 is mediated with the induction of DR4/DR5, Egr-1 and NAG-1, activation of caspases and modulation of the Bcl-2 family in p53-deficient Hep3B hepatocellular carcinoma cells. Oncol. Rep. 2008, 19, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J. Early Growth Response Gene Upregulation in Epstein-Barr Virus (EBV)-Associated Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Biomolecules 2020, 10, 1484. [Google Scholar] [CrossRef]
- Dyson, O.F.; Traylen, C.M.; Akula, S.M. Cell membrane-bound Kaposi’s sarcoma-associated herpesvirus-encoded glycoprotein B promotes virus latency by regulating expression of cellular Egr-1. J. Biol. Chem. 2010, 285, 37491–37502. [Google Scholar] [CrossRef] [Green Version]
- Buehler, J.; Carpenter, E.; Zeltzer, S.; Igarashi, S.; Rak, M.; Mikell, I.; Nelson, J.A.; Goodrum, F. Host signaling and EGR1 transcriptional control of human cytomegalovirus replication and latency. PLoS Pathog. 2019, 15, e1008037. [Google Scholar] [CrossRef]
- Zhu, Z.; Du, X.; Li, P.; Zhang, X.; Yang, F.; Cao, W.; Tian, H.; Zhang, K.; Liu, X.; Zheng, H. Early Growth Response Gene-1 Suppresses Foot-and-Mouth Disease Virus Replication by Enhancing Type I Interferon Pathway Signal Transduction. Front. Microbiol. 2018, 9, 2326. [Google Scholar] [CrossRef]
- Dahal, B.; Lin, S.-C.; Carey, B.D.; Jacobs, J.L.; Dinman, J.; van Hoek, M.; Adams, A.A.; Kehn-Hall, K. EGR1 upregulation following Venezuelan equine encephalitis virus infection is regulated by ERK and PERK pathways contributing to cell death. Virology 2020, 539, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Matys, V. TRANSFAC(R) and its module TRANSCompel(R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendra, J.A.; de la Fuente, C.; Brahms, A.; Woodson, C.; Bell, T.M.; Chen, B.; Khan, Y.; Jacobs, J.L.; Kehn-Hall, K.; Dinman, J.D. Ablation of Programmed −1 Ribosomal Frameshifting in Venezuelan Equine Encephalitis Virus Results in Attenuated Neuropathogenicity. J. Virol. 2017, 91, e01766-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, A.; Bansal, N.; Senina, S.; Hooper, I.; Lundberg, L.; De La Fuente, C.; Narayanan, A.; Gutting, B.W.; Kehn-Hall, K. Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Front. Microbiol. 2015, 6, 676. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, L.; Pinkham, C.; de la Fuente, C.; Brahms, A.; Shafagati, N.; Wagstaff, K.M.; Jans, D.A.; Tamir, S.; Kehn-Hall, K. Selective Inhibitor of Nuclear Export (SINE) Compounds Alter New World Alphavirus Capsid Localization and Reduce Viral Replication in Mammalian Cells. PLoS Negl. Trop. Dis. 2016, 10, e0005122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.; Kehn-Hall, K. Viral concentration determination through plaque assays: Using traditional and novel overlay systems. J. Vis. Exp. 2014, 93, e52065. [Google Scholar] [CrossRef] [PubMed]
- Salimi, H.; Cain, M.D.; Klein, R.S. Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics 2016, 13, 514–534. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Herrero, L.J.; Rudd, P.A.; Mahalingam, S. Mouse models of alphavirus-induced inflammatory disease. J. Gen. Virol. 2015, 96 Pt 2, 221–238. [Google Scholar] [CrossRef]
- Mechtcheriakova, D.; Schabbauer, G.; Lucerna, M.; Clauss, M.; De Martin, R.; Binder, B.R.; Hofer, E. Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor up-regulation via EGR-1 in endothelial cells. FASEB J. 2001, 15, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Decker, E.L.; Nehmann, N.; Kampen, E.; Eibel, H.; Zipfel, P.F.; Skerka, C. Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. 2003, 31, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Bhattacharya, B.; Puri, R.K.; Maheshwari, R.K. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 2008, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Nicoll, M.; Ingham, R.J. AP-1 family transcription factors: A diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL. Exp. Hematol. Oncol. 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-K.; Wu, H.-C.; Shen, Y.-C.; Hsieh, H.-Y.; Yang, S.-Y.; Chang, C.-C. Krüppel-like factor 4 is involved in cell scattering induced by hepatocyte growth factor. J. Cell Sci. 2012, 125 Pt 20, 4853–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Maheshwari, R.K. Oligonucleotide array analysis of Toll-like receptors and associated signalling genes in Venezuelan equine encephalitis virus-infected mouse brain. J. Gen. Virol. 2009, 90 Pt 8, 1836–1847. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kanneganti, T.-D. Caspase-7: A protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Reyes, N.; Figueroa, S.; Tiwari, R.; Geliebter, J. CXCL3 Signaling in the Tumor Microenvironment. Single Mol. Single Cell Seq. 2021, 1302, 15–24. [Google Scholar]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Rajarathnam, K.; Schnoor, M.; Richardson, R.M.; Rajagopal, S. How do chemokines navigate neutrophils to the target site: Dissecting the structural mechanisms and signaling pathways. Cell. Signal. 2018, 54, 69–80. [Google Scholar] [CrossRef]
- Skinner, D.; Marro, B.; Lane, T.E. Chemokine CXCL10 and Coronavirus-Induced Neurologic Disease. Viral Immunol. 2019, 32, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.S. Role and regulation of cyclooxygenase-2 during inflammation. Am. J. Med. 1999, 106, 37S–42S. [Google Scholar] [CrossRef]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Xu, D.; Williams, B.R.G. ATF3 transcription factor and its emerging roles in immunity and cancer. Klin. Wochenschr. 2009, 87, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Liu, Z.-G.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Poirier, R.; Cheval, H.; Mailhes, C.; Garel, S.; Charnay, P.; Davis, S.; Laroche, S. Distinct functions of Egr gene family members in cognitive processes. Front. Neurosci. 2008, 2, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska, M.; Juzwa, W.; Krzyzosiak, W.J.; Olejniczak, M. Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front. Neurosci. 2018, 12, 75. [Google Scholar] [CrossRef]
- Swanson, P.A., II; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, A.L.; Powell, D.S.; Bethel, L.M.; Caroline, A.L.; Schmid, R.J.; Oury, T.; Reed, D.S. Aerosolized Rift Valley Fever Virus Causes Fatal Encephalitis in African Green Monkeys and Common Marmosets. J. Virol. 2014, 88, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, P.M.; Andreadis, T.G. Ecology and Epidemiology of Eastern Equine Encephalitis Virus in the Northeastern United States: An Historical Perspective. J. Med. Entomol. 2021, 59, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ciota, A.T. Eastern Equine Encephalitis Virus Taxonomy, Genomics, and Evolution. J. Med. Entomol. 2021, 59, 14–19. [Google Scholar] [CrossRef]
- Corrin, T.; Ackford, R.; Mascarenhas, M.; Greig, J.; Waddell, L.A. Eastern Equine Encephalitis Virus: A Scoping Review of the Global Evidence. Vector-Borne Zoonotic Dis. 2021, 21, 305–320. [Google Scholar] [CrossRef]
- Atkins, G.J.; Sheahan, B.J. Molecular determinants of alphavirus neuropathogenesis in mice. J. Gen. Virol. 2016, 97, 1283–1296. [Google Scholar] [CrossRef] [Green Version]
- Costa, B.K.D.; Sato, D.K. Viral encephalitis: A practical review on diagnostic approach and treatment. J. Pediatr. 2020, 96 (Suppl. 1), 12–19. [Google Scholar] [CrossRef]
- Doughty, C.T.; Yawetz, S.; Lyons, J. Emerging Causes of Arbovirus Encephalitis in North America: Powassan, Chikungunya, and Zika Viruses. Curr. Neurol. Neurosci. Rep. 2017, 17, 12. [Google Scholar] [CrossRef]
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar] [CrossRef]
- Irusta, P.M.; Hardwick, J.M. Neuronal Apoptosis Pathways in Sindbis Virus Encephalitis. Prog. Mol. Subcell. Biol. 2004, 36, 71–93. [Google Scholar]
- Hartman, A. Rift Valley Fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Hirayama, D.; Wagatsuma, K.; Yamakawa, T.; Yokoyama, Y.; Nakase, H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 3062. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-M.; Luo, W.; Wang, H.; Li, R.-Z.; Huang, Y.-S.; Chen, L.-K.; Wu, X.-P. The Role of Prostaglandin-Endoperoxide Synthase-2 in Chemoresistance of Non-Small Cell Lung Cancer. Front. Pharmacol. 2019, 10, 836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Zanini, F.; Kumar, S.; Karim, M.; Saul, S.; Bhalla, N.; Panpradist, N.; Muniz, A.; Narayanan, A.; Quake, S.R.; et al. The transcriptional landscape of Venezuelan equine encephalitis virus (TC-83) infection. PLoS Negl. Trop. Dis. 2021, 15, e0009306. [Google Scholar] [CrossRef]
- Luo, W.-W.; Lian, H.; Zhong, B.; Shu, H.-B.; Li, S. Krüppel-like factor 4 negatively regulates cellular antiviral immune response. Cell. Mol. Immunol. 2014, 13, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, N.C.; Bahn, J.H.; Lee, S.-H.; Eling, T.E.; Baek, S.J. Resveratrol-Induced Apoptosis Is Mediated by Early Growth Response-1, Krüppel-Like Factor 4, and Activating Transcription Factor 3. Cancer Prev. Res. 2011, 4, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Hong, Y.; Zhan, Q.; Shen, Y.; Liu, Z. Role for Krüppel-Like Factor 4 in Determining the Outcome of p53 Response to DNA Damage. Cancer Res. 2009, 69, 8284–8292. [Google Scholar] [CrossRef] [Green Version]
- Michlmayr, D.; Lim, J.K. Chemokine receptors as important regulators of pathogenesis during arboviral encephalitis. Front. Cell Neurosci. 2014, 8, 264. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Schoneboom, B.A.; Catlin, K.M.; Marty, A.M.; Grieder, F.B. Inflammation is a component of neurodegeneration in response to Venezuelan equine encephalitis virus infection in mice. J. Neuroimmunol. 2000, 109, 132–146. [Google Scholar] [CrossRef]
- Chirathaworn, C.; Chansaenroj, J.; Poovorawan, Y. Cytokines and Chemokines in Chikungunya Virus Infection: Protection or Induction of Pathology. Pathogens 2020, 9, 415. [Google Scholar] [CrossRef]
- Fox, J.M.; Diamond, M.S. Immune-Mediated Protection and Pathogenesis of Chikungunya Virus. J. Immunol. 2016, 197, 4210–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prow, N.A.; Hirata, T.D.C.; Tang, B.; Larcher, T.; Mukhopadhyay, P.; Alves, T.L.; Le, T.T.; Gardner, J.; Poo, Y.S.; Nakayama, E.; et al. Exacerbation of Chikungunya Virus Rheumatic Immunopathology by a High Fiber Diet and Butyrate. Front. Immunol. 2019, 10, 2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekchariyawat, P.; Hamel, R.; Bernard, E.; Wichit, S.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; Choumet, V.; Yssel, H.; Desprès, P.; et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 2015, 32, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Baxter, V.K.; Griffin, D.E. Interferon gamma modulation of disease manifestation and the local antibody response to alphavirus encephalomyelitis. J. Gen. Virol. 2016, 97, 2908–2925. [Google Scholar] [CrossRef]
- Metcalf, T.U.; Baxter, V.K.; Nilaratanakul, V.; Griffin, D.E. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J. Virol. 2013, 87, 2420–2429. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.A.; Sgarioni, E.; Boquett, J.A.; Terças-Trettel, A.C.P.; da Silva, J.H.; Ribeiro, B.F.R.; Galera, M.F.; de Oliveira, T.M.; Carvalho de Andrade, M.D.F.; Carvalho, I.F.; et al. Association between Genetic Variants in NOS2 and TNF Genes with Congenital Zika Syndrome and Severe Microcephaly. Viruses 2021, 13, 325. [Google Scholar] [CrossRef]
- de St Maurice, A.; Harmon, J.; Nyakarahuka, L.; Balinandi, S.; Tumusiime, A.; Kyondo, J.; Mulei, S.; Namutebi, A.; Knust, B.; Spiropoulou, C.F.; et al. Rift valley fever viral load correlates with the human inflammatory response and coagulation pathway abnormalities in humans with hemorrhagic manifestations. PLoS Negl. Trop. Dis. 2018, 12, e0006460. [Google Scholar] [CrossRef] [Green Version]
- Jansen van Vuren, P.; Tiemessen, C.T.; Paweska, J.T. Anti-nucleocapsid protein immune responses counteract pathogenic effects of Rift Valley fever virus infection in mice. PLoS ONE 2011, 6, e25027. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Puljak, L.; Marin, A.; Vrdoljak, D.; Markotic, F.; Utrobicic, A.; Tugwell, P. Celecoxib for osteoarthritis. Cochrane Database Syst. Rev. 2017, 5, CD009865. [Google Scholar] [CrossRef] [PubMed]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of FDA-Approved Anti-Inflammatory Drugs Against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Sequence |
|---|---|
| Guide RNA target sequence-located in Exon 1 of EGR1 | 5′ ctttcctcactcgcccacca 3′ |
| EGR1 Exon 1 Fwd | 5′ gagagatccagccgcagaac 3′ |
| EGR1 Exon 1 Rev | 5′ cggtcaggtgctcgtaggg 3′ |
| Gene | Entrez Gene Name | Log2 Fold Change 1 | p-Value 1 | FDR p-Value 1 |
|---|---|---|---|---|
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit | 3.510 | 2.67 × 10−97 | 3.37 × 10−94 |
| CXCL3 | C-X-C motif chemokine ligand 3 | 3.225 | 7.30 × 10−4 | 0.03 |
| CXCL8 | C-X-C motif chemokine ligand 8 | 2.556 | 0.02 | 0.39 |
| CASP7 | Caspase 7 | 1.269 | 0.03 | 0.47 |
| SERPINE1 | Serpin family E member 1 | 2.170 | 1.21 × 10−90 | 1.41 × 10−87 |
| SNAI2 | Snail family transcriptional repressor 2 | 1.740 | 2.05 × 10−4 | 9.67 × 10−3 |
| FTL | Ferritin Light Chain | 0.556 | 5.35 × 10−5 | 3.29 × 10−3 |
| CLU | Clusterin | 1.316 | 7.56 × 10−11 | 1.52 × 10−8 |
| FOSL1 | FOS-like 1, AP-1 transcription factor subunit | 1.411 | 3.9 × 10−9 | 6.02 × 10−7 |
| CD44 | CD44 Molecule | 0.651 | 5.86 × 10−4 | 0.02 |
| HMOX1 | Heme oxygenase 1 | 3.076 | 3.58 × 10−39 | 2.37 × 10−36 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | 5.525 | 4.79 × 10−7 | 5.37 × 10−5 |
| ATF3 | Activating transcription factor 3 | 5.260 | 2.36 × 10−40 | 1.64 × 10−37 |
| EGR1 | Early growth response 1 | 5.841 | 1.58 × 10−81 | 1.56 × 10−78 |
| Gene | Entrez Gene Name | Function | Log2 Fold Change 1 |
|---|---|---|---|
| CASP7 | Caspase 7 | Involved in the activation cascade of caspases responsible for apoptosis execution [37]. | 1.269 |
| CXCL3 | C-X-C Motif Chemokine Ligand 3 | Chemokine involved in inflammation; chemoattractant for neutrophils [38]. | 3.225 |
| CXCL8 | C-X-C Motif Chemokine Ligand 8 | Major mediator of the inflammatory response. Functions as a chemotactic factor by guiding the neutrophils to the site of infection [39,40]. | 2.556 |
| CXCL10 | C-X-C Motif Chemokine Ligand 10 | Stimulates production of monocytes, natural killer and T-cell migration, and modulation of adhesion molecule expression [41]. | N/A * |
| TNF-α | Tumor Necrosis Factor Alpha | Pro-inflammatory cytokine involved in regulation of a wide variety of biological processes, including apoptosis [42]. | 7.415 |
| PTGS2 | Prostaglandin-Endoperoxide Synthase 2 | Key enzyme in prostaglandin biosynthesis: a group of lipids made at sites of tissue damage or infection that are involved in dealing with injury and illness [43]. | 5.525 |
| TGF-β | Transforming growth factor beta (TGF-β) | Multifunctional cytokine. Plays a role in immune and stem cell regulation and differentiation [44]. | 0.447 |
| Gene | Entrez Gene Name | Function | Log2 Fold Change 1 |
|---|---|---|---|
| ATF3 | Activating transcription factor 3 | Binds the cAMP response element (CRE) (consensus: 5′-GTGACGT[AC][AG]-3′), a sequence present in many viral and cellular promoters. Plays a role in regulating the cell cycle and apoptosis [45]. | 5.260 |
| FOS | Fos Proto-Oncogene | Dimerizes with proteins of the JUN family, thereby forming the transcription factor complex AP-1. Involved in regulation of cell proliferation, differentiation, transformation, and apoptosis [46]. | 3.387 |
| JUN | Jun Proto-Oncogene | Second factor of AP-1 transcription factor complex. Interacts with specific target DNA sequences to regulate gene expression [47]. | 3.510 |
| KLF4 | Kruppel-Like Factor 4 | Thought to control the G1-to-S transition of the cell cycle following DNA damage by mediating the tumor suppressor gene p53 [48]. | 4.360 |
| Virus | Family, Genus | Known to Cause Encephalitis? |
|---|---|---|
| Eastern equine encephalitis virus | Togaviridae, Alphavirus (New World) | Yes [53,54,55] |
| Chikungunya virus | Togaviridae, Alphavirus (Old World) | Sometimes [56,57,58,59] |
| Sindbis virus | Togaviridae, Alphavirus (Old World) | Sometimes [56,60] |
| Zika virus | Flaviviridae, Flavivirus | Yes [57,58,59] |
| Rift Valley fever virus | Phenuiviridae, Phlebovirus | Yes [52,59,61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehman, C.W.; Smith, A.; Kelly, J.; Jacobs, J.L.; Dinman, J.D.; Kehn-Hall, K. EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death. Viruses 2022, 14, 1210. https://doi.org/10.3390/v14061210
Lehman CW, Smith A, Kelly J, Jacobs JL, Dinman JD, Kehn-Hall K. EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death. Viruses. 2022; 14(6):1210. https://doi.org/10.3390/v14061210
Chicago/Turabian StyleLehman, Caitlin W., Amy Smith, Jamie Kelly, Jonathan L. Jacobs, Jonathan D. Dinman, and Kylene Kehn-Hall. 2022. "EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death" Viruses 14, no. 6: 1210. https://doi.org/10.3390/v14061210
APA StyleLehman, C. W., Smith, A., Kelly, J., Jacobs, J. L., Dinman, J. D., & Kehn-Hall, K. (2022). EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death. Viruses, 14(6), 1210. https://doi.org/10.3390/v14061210