
Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice


Abstract
:1. Introduction
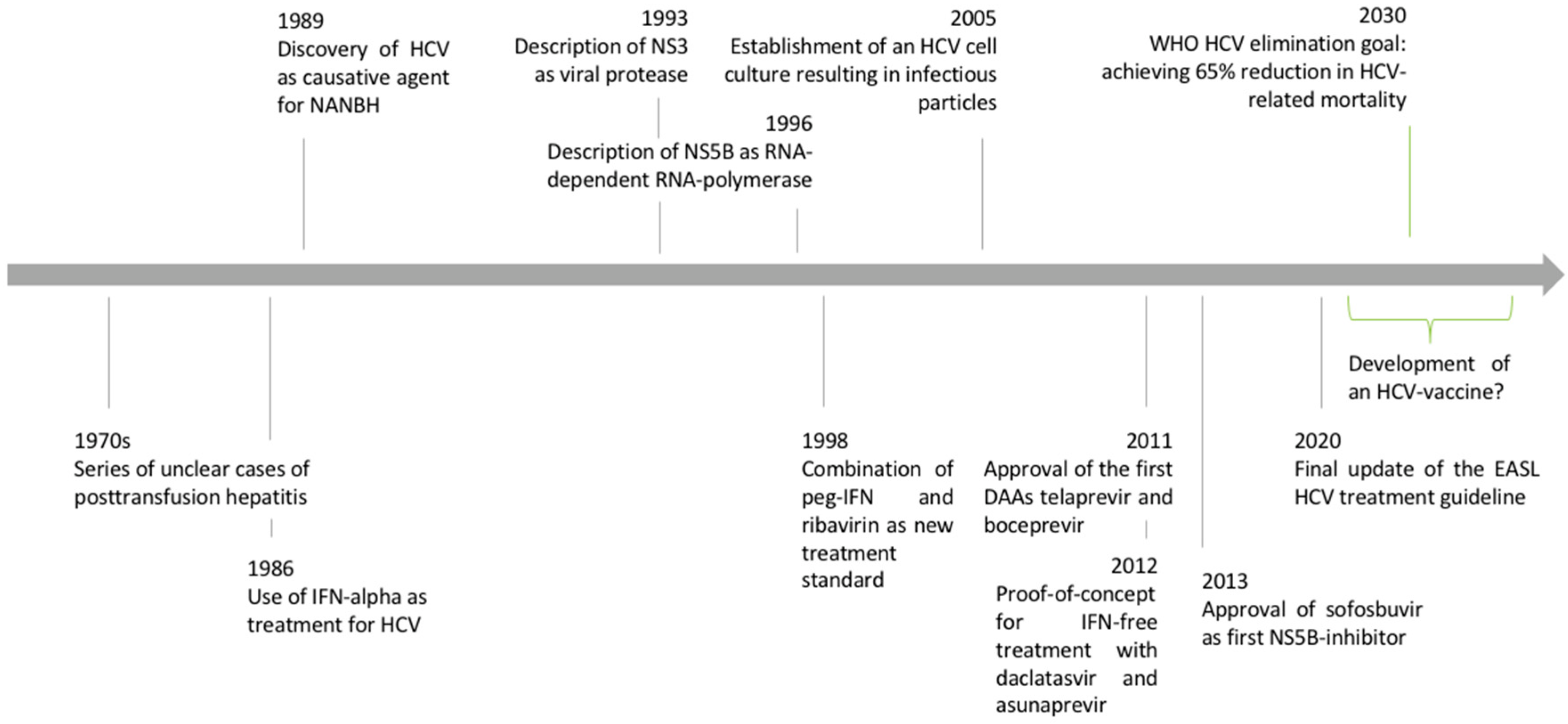
1.1. Discovery of the Hepatitis C Virus
1.2. Epidemiology and Natural History of Hepatitis C Virus Infection
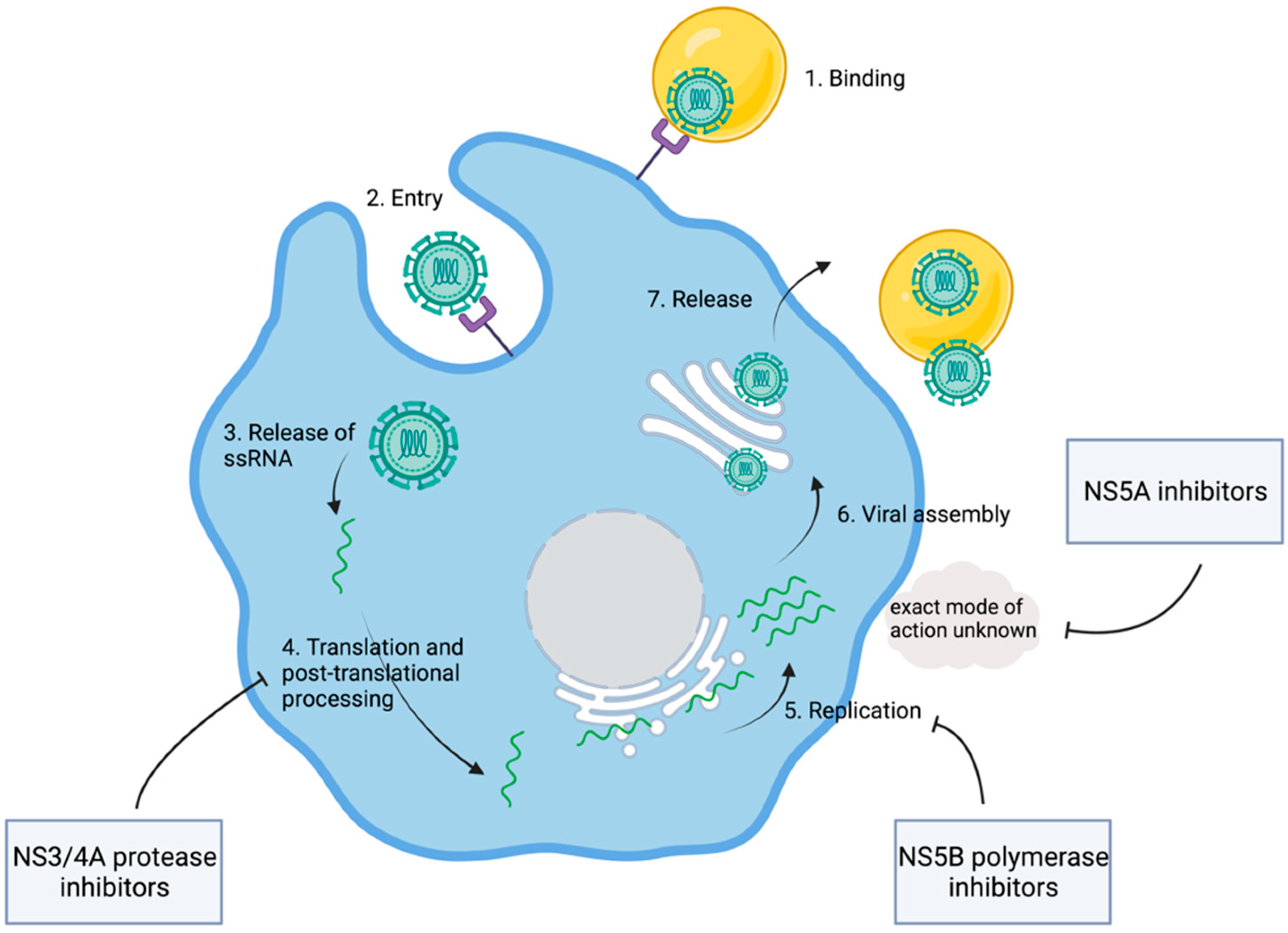
1.3. Discovery of the HCV Replication Cycle
2. Development of Direct-Acting Antiviral Agents
2.1. Sofosbuvir/Velpatasvir (SOF/VEL)
2.2. Glecaprevir/Pibrentasvir (G/P)
2.3. Grazoprevir/Elbasvir
2.4. Sofosbuvir/Velpatasvir/Voxilaprevir (SOF/VEL/VOX)
2.5. Current Treatment Strategies
2.6. Developing an HCV Vaccination—A Remaining Challenge
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghany, M.G.; Lok, A.S.F.; Dienstag, J.L.; Feinstone, S.M.; Hoofnagle, J.H.; Jake Liang, T.; Seeff, L.B.; Cohen, D.E.; Bezerra, J.A.; Chung, R.T. The 2020 Nobel Prize for Medicine or Physiology for the Discovery of Hepatitis C Virus: A Triumph of Curiosity and Persistence. Hepatology 2021, 74, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.; Maasoumy, B. Breakthroughs in hepatitis C research: From discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 2022, 11, 2565. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-Associated Hepatitis Not Due to Viral Hepatitis Type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isol. a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, G.; Choo, Q.L.; Alter, H.J.; Gitnick, G.L.; Redeker, A.G.; Purcell, R.H.; Miyamura, T.; Dienstag, J.L.; Alter, M.J.; Stevens, C.E.; et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 1989, 244, 362–366. [Google Scholar] [CrossRef]
- Bertoletti, A.; Ferrari, C. Kinetics of the immune response during HBV and HCV infection. Hepatology 2003, 38, 4–13. [Google Scholar] [CrossRef]
- Maasoumy, B.; Wedemeyer, H. Natural history of acute and chronic hepatitis C. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 401–412. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef]
- Cacoub, P.; Gragnani, L.; Comarmond, C.; Zignego, A.L. Extrahepatic manifestations of chronic hepatitis C virus infection. Dig. Liver Dis. 2014, 46, S165–S173. [Google Scholar] [CrossRef] [Green Version]
- Larney, S.; Peacock, A.; Leung, J.; Colledge, S.; Hickman, M.; Vickerman, P.; Grebely, J.; Dumchev, K.V.; Griffiths, P.; Hines, L.; et al. Global, regional, and country-level coverage of interventions to prevent and manage HIV and hepatitis C among people who inject drugs: A systematic review. Lancet Glob. Health 2017, 5, e1208–e1220. [Google Scholar] [CrossRef] [Green Version]
- Grebely, J.; Prins, M.; Hellard, M.; Cox, A.L.; Osburn, W.O.; Lauer, G.; Page, K.; Lloyd, A.R.; Dore, G.J. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: Towards a vaccine. Lancet Infect. Dis. 2012, 12, 408–414. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections, 2021. Available online: https://www.who.int/publications/i/item/9789240027077 (accessed on 4 March 2022).
- WHO. Global Health Sector Strategy on Viral Hepatitis 2016–2021–Towards Ending Viral Hepatitis. 2016. Available online: https://apps.who.int/iris/handle/10665/246177 (accessed on 4 March 2022).
- Lohmann, V. Hepatitis C virus RNA replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar] [PubMed]
- Takamizawa, A.; Mori, C.; Fuke, I.; Manabe, S.; Murakami, S.; Fujita, J.; Onishi, E.; Andoh, T.; Yoshida, I.; Okayama, H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 1991, 65, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, Q.L.; Richman, K.H.; Han, J.H.; Berger, K.; Lee, C.; Dong, C.; Gallegos, C.; Coit, D.; Medina-Selby, R.; Barr, P.J.; et al. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 2451–2455. [Google Scholar] [CrossRef] [Green Version]
- Hijikata, M.; Kato, N.; Ootsuyama, Y.; Nakagawa, M.; Shimotohno, K. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 1991, 88, 5547–5551. [Google Scholar] [CrossRef] [Green Version]
- Grakoui, A.; Wychowski, C.; Lin, C.; Feinstone, S.M.; Rice, C.M. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 1993, 67, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol. 1993, 67, 3835–4384. [Google Scholar] [CrossRef] [Green Version]
- Failla, C.; Tomei, L.; De Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 1994, 68, 3753–3760. [Google Scholar] [CrossRef] [Green Version]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 1994, 68, 5045–5055. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. USA 1990, 87, 2057–2061. [Google Scholar] [CrossRef] [Green Version]
- Behrens, S.E.; Tomei, L.; De Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef] [Green Version]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.J.; Von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; De Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, V.; Körner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Lamarre, D.; Anderson, P.C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bös, M.; Cameron, D.R.; Cartier, M.; Cordingley, M.G.; et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [Google Scholar] [CrossRef]
- Hinrichsen, H.; Benhamou, Y.; Wedemeyer, H.; Reiser, M.; Sentjens, R.E.; Calleja, J.L.; Forns, X.; Erhardt, A.; Cronlein, J.; Chaves, R.L.; et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 2004, 127, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Reiser, M.; Hinrichsen, H.; Benhamou, Y.; Reesink, H.W.; Wedemeyer, H.; Avendano, C.; Riba, N.; Yong, C.L.; Nehmiz, G.; Steinmann, G.G. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology 2005, 41, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Vanwolleghem, T.; Meuleman, P.; Libbrecht, L.; Roskams, T.; De Vos, R.; Leroux-Roels, G. Ultra-rapid cardiotoxicity of the hepatitis C virus protease inhibitor BILN 2061 in the urokinase-type plasminogen activator mouse. Gastroenterology 2007, 133, 1144–1155. [Google Scholar] [CrossRef]
- Sherman, K.E.; Flamm, S.L.; Afdhal, N.H.; Nelson, D.R.; Sulkowski, M.S.; Everson, G.T.; Fried, M.W.; Adler, M.; Reesink, H.W.; Martin, M.; et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 2011, 365, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHutchison, J.G.; Manns, M.P.; Muir, A.J.; Terrault, N.A.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.J.; Zeuzem, S.; Reesink, H.W.; Garg, J.; et al. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 2010, 362, 1292–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeuzem, S.; Andreone, P.; Pol, S.; Lawitz, E.; Diago, M.; Roberts, S.; Focaccia, R.; Younossi, Z.; Foster, G.R.; Horban, A.; et al. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 2011, 364, 2417–2428. [Google Scholar] [CrossRef] [Green Version]
- Poordad, F.; McCone, J., Jr.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef] [Green Version]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef] [Green Version]
- Jesudian, A.B.; Jacobson, I.M. Optimal treatment with telaprevir for chronic HCV infection. Liver Int. 2013, 33, 3–13. [Google Scholar] [CrossRef]
- Vierling, J.M.; Zeuzem, S.; Poordad, F.; Bronowicki, J.P.; Manns, M.P.; Bacon, B.R.; Esteban, R.; Flamm, S.L.; Kwo, P.Y.; Pedicone, L.D.; et al. Safety and efficacy of boceprevir/peginterferon/ribavirin for HCV G1 compensated cirrhotics: Meta-analysis of 5 trials. J. Hepatol. 2014, 61, 200–209. [Google Scholar] [CrossRef]
- Maasoumy, B.; Port, K.; Markova, A.A.; Serrano, B.C.; Rogalska-Taranta, M.; Sollik, L.; Mix, C.; Kirschner, J.; Manns, M.P.; Wedemeyer, H.; et al. Eligibility and safety of triple therapy for hepatitis C: Lessons learned from the first experience in a real world setting. PLoS ONE 2013, 8, e55285. [Google Scholar]
- Manns, M.P.; Cornberg, M. Sofosbuvir: The final nail in the coffin for hepatitis C? Lancet Infect. Dis. 2013, 13, 378–379. [Google Scholar] [CrossRef]
- Silva, M.O.; Treitel, M.; Graham, D.J.; Curry, S.; Frontera, M.J.; McMonagle, P.; Gupta, S.; Hughes, E.; Chase, R.; Lahser, F.; et al. Antiviral activity of boceprevir monotherapy in treatment-naive subjects with chronic hepatitis C genotype 2/3. J. Hepatol. 2013, 59, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.G.; Sablon, E.; Chamberland, J.; Fournier, E.; Dandavino, R.; Tremblay, C.L. Hepatitis C virus genotype 7, a new genotype originating from central Africa. J. Clin. Microbiol. 2015, 53, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Lemm, J.A.; O’Boyle, D., 2nd; Liu, M.; Nower, P.T.; Colonno, R.; Deshpande, M.S.; Snyder, L.B.; Martin, S.W.; St Laurent, D.R.; Serrano-Wu, M.H.; et al. Identification of hepatitis C virus NS5A inhibitors. J. Virol. 2010, 84, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Manns, M.; Pol, S.; Jacobson, I.M.; Marcellin, P.; Gordon, S.C.; Peng, C.Y.; Chang, T.T.; Everson, G.T.; Heo, J.; Gerken, G.; et al. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: A multinational, phase 3, multicohort study. Lancet 2014, 384, 1597–1605. [Google Scholar] [CrossRef]
- Ludmerer, S.W.; Graham, D.J.; Boots, E.; Murray, E.M.; Simcoe, A.; Markel, E.J.; Grobler, J.A.; Flores, O.A.; Olsen, D.B.; Hazuda, D.J.; et al. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob. Agents Chemother. 2005, 49, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Abraham, G.M.; Spooner, L.M. Sofosbuvir in the treatment of chronic hepatitis C: New dog, new tricks. Clin. Infect. Dis. 2014, 59, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honer Zu Siederdissen, C.; Maasoumy, B.; Marra, F.; Deterding, K.; Port, K.; Manns, M.P.; Cornberg, M.; Back, D.; Wedemeyer, H. Drug-Drug Interactions with Novel All Oral Interferon-Free Antiviral Agents in a Large Real-World Cohort. Clin. Infect. Dis. 2016, 62, 561–567. [Google Scholar] [CrossRef]
- Puoti, M.; Panzeri, C.; Rossotti, R.; Baiguera, C. Efficacy of sofosbuvir-based therapies in HIV/HCV infected patients and persons who inject drugs. Dig. Liver Dis. 2014, 46, S206–S211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, M.S.; Naggie, S.; Lalezari, J.; Fessel, W.J.; Mounzer, K.; Shuhart, M.; Luetkemeyer, A.F.; Asmuth, D.; Gaggar, A.; Ni, L.; et al. Sofosbuvir and ribavirin for hepatitis C in patients with HIV coinfection. JAMA 2014, 312, 353–361. [Google Scholar] [CrossRef] [Green Version]
- EASL Recommendations on Treatment of Hepatitis C: Final Update of the Series. J. Hepatol. 2020, 73, 1170–1218. [CrossRef]
- Kirby, B.J.; Symonds, W.T.; Kearney, B.P.; Mathias, A.A. Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of the Hepatitis C Virus NS5B Polymerase Inhibitor Sofosbuvir. Clin. Pharmacokinet. 2015, 54, 677–690. [Google Scholar] [CrossRef]
- Mogalian, E.; German, P.; Kearney, B.P.; Yang, C.Y.; Brainard, D.; McNally, J.; Moorehead, L.; Mathias, A. Use of Multiple Probes to Assess. Transporter- and Cytochrome P450-Mediated Drug-Drug Interaction Potential of the Pangenotypic HCV NS5A Inhibitor Velpatasvir. Clin. Pharmacokinet. 2016, 55, 605–613. [Google Scholar] [CrossRef]
- Feld, J.J.; Jacobson, I.M.; Hezode, C.; Asselah, T.; Ruane, P.J.; Gruener, N.; Abergel, A.; Mangia, A.; Lai, C.L.; Chan, H.L.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection. N. Engl. J. Med. 2015, 333, 2599–2607. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R.; Afdhal, N.; Roberts, S.K.; Brau, N.; Gane, E.J.; Pianko, S.; Lawitz, E.; Thompson, A.; Shiffman, M.L.; Cooper, C.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. N. Engl. J. Med. 2015, 373, 2608–2617. [Google Scholar] [CrossRef] [Green Version]
- Curry, M.P.; O’Leary, J.G.; Bzowej, N.; Muir, A.J.; Korenblat, K.M.; Fenkel, J.M.; Reddy, K.R.; Lawitz, E.; Flamm, S.L.; Schiano, T.; et al. Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis. N. Engl. J. Med. 2015, 373, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.; Brau, N.; Kottilil, S.; Daar, E.S.; Ruane, P.; Workowski, K.; Luetkemeyer, A.; Adeyemi, O.; Kim, A.Y.; Doehle, B.; et al. Sofosbuvir and Velpatasvir for the Treatment of Hepatitis C Virus in Patients Coinfected with Human Immunodeficiency Virus Type 1: An Open-Label, Phase 3 Study. Clin. Infect. Dis. 2017, 65, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puoti, M.; Foster, G.R.; Wang, S.; Mutimer, D.; Gane, E.; Moreno, C.; Chang, T.T.; Lee, S.S.; Marinho, R.; Dufour, J.F.; et al. High SVR12 with 8-week and 12-week glecaprevir/pibrentasvir therapy: An integrated analysis of HCV genotype 1–6 patients without cirrhosis. J. Hepatol. 2018, 69, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosio, R.; Pasulo, L.; Puoti, M.; Vinci, M.; Schiavini, M.; Lazzaroni, S.; Soria, A.; Gatti, F.; Menzaghi, B.; Aghemo, A.; et al. Real-world effectiveness and safety of glecaprevir/pibrentasvir in 723 patients with chronic hepatitis C. J. Hepatol. 2019, 70, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Naumann, U.; Stoehr, A.; Sick, C.; John, C.; Teuber, G.; Schiffelholz, W.; Mauss, S.; Lohmann, K.; König, B.; et al. Real-world effectiveness and safety of glecaprevir/pibrentasvir for the treatment of chronic hepatitis C infection: Data from the German Hepatitis C-Registry. Aliment. Pharmacol. Ther. 2019, 49, 1052–1059. [Google Scholar] [CrossRef]
- Zeuzem, S.; Serfaty, L.; Vierling, J.; Cheng, W.; George, J.; Sperl, J.; Strasser, S.; Kumada, H.; Hwang, P.; Robertson, M.; et al. The safety and efficacy of elbasvir and grazoprevir in participants with hepatitis C virus genotype 1b infection. J. Gastroenterol. 2018, 53, 679–688. [Google Scholar] [CrossRef]
- Jacobson, I.M.; Lawitz, E.; Kwo, P.Y.; Hezode, C.; Peng, C.Y.; Howe, A.Y.M.; Hwang, P.; Wahl, J.; Robertson, M.; Barr, E.; et al. Safety and Efficacy of Elbasvir/Grazoprevir in Patients with Hepatitis C Virus Infection and Compensated Cirrhosis: An Integrated Analysis. Gastroenterology 2017, 152, 1372–1382.e2. [Google Scholar] [CrossRef] [Green Version]
- Bourliere, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef]
- Belperio, P.S.; Shahoumian, T.A.; Loomis, T.P.; Backus, L.I. Real-world effectiveness of sofosbuvir/velpatasvir/voxilaprevir in 573 direct-acting antiviral experienced hepatitis C patients. J. Viral Hepat. 2019, 26, 980–990. [Google Scholar] [CrossRef]
- Degasperi, E.; Spinetti, A.; Lombardi, A.; Landonio, S.; Rossi, M.C.; Pasulo, L.; Pozzoni, P.; Giorgini, A.; Fabris, P.; Romano, A.; et al. Real-life effectiveness and safety of sofosbuvir/velpatasvir/voxilaprevir in hepatitis C patients with previous DAA failure. J. Hepatol. 2019, 71, 1106–1115. [Google Scholar] [CrossRef]
- Foster, G.R.; Irving, W.L.; Cheung, M.C.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.; et al. Impact of direct acting antiviral therapy in patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 64, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Belli, L.S.; Berenguer, M.; Cortesi, P.A.; Strazzabosco, M.; Rockenschaub, S.R.; Martini, S.; Morelli, C.; Donato, F.; Volpes, R.; Pageaux, G.P.; et al. Delisting of liver transplant candidates with chronic hepatitis C after viral eradication: A European study. J. Hepatol. 2016, 65, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Crespo, G.; Trota, N.; Londoño, M.C.; Mauro, E.; Baliellas, C.; Castells, L.; Castellote, J.; Tort, J.; Forns, X.; Navasa, M. The efficacy of direct anti-HCV drugs improves early post-liver transplant survival and induces significant changes in waiting list composition. J. Hepatol. 2018, 69, 11–17. [Google Scholar] [CrossRef]
- Waked, I.; Esmat, G.; Elsharkawy, A.; El-Serafy, M.; Abdel-Razek, W.; Ghalab, R.; Elshishiney, G.; Salah, A.; Abdel Megid, S.; Kabil, K.; et al. Screening and Treatment Program to Eliminate Hepatitis C in Egypt. N. Engl. J. Med. 2020, 382, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-L.; Chang, M.-H.; Ni, Y.-H.; Hsu, H.-Y.; Lee, P.-l.; Lee, C.-Y.; Chen, D.-S. Seroepidemiology of Hepatitis B Virus Infection in Children: Ten Years of Mass Vaccination in Taiwan. JAMA 1996, 276, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef]
- Page, K.; Melia, M.T.; Veenhuis, R.T.; Winter, M.; Rousseau, K.E.; Massaccesi, G.; Osburn, W.O.; Forman, M.; Thomas, E.; Thornton, K.; et al. Randomized Trial of a Vaccine Regimen to Prevent Chronic HCV Infection. N. Engl. J. Med. 2021, 384, 541–549. [Google Scholar] [CrossRef]
- Sepulveda-Crespo, D.; Resino, S.; Martinez, I. Hepatitis C virus vaccine design: Focus on the humoral immune response. J. Biomed. Sci. 2020, 27, 78. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Nonstructural Proteins | Molecular Function | Drug Targeting |
|---|---|---|
| NS3 | Viral protease cleavage between NS3/NS4 and NS4/NS5 | Protease inhibitors, e.g., glecaprevir, voxilaprevir, grazoprevir |
| NS4A | Cofactor in viral proteolytic activity of NS3 | / |
| NS5A | Mediates interactions between viral and host proteins | NS5A inhibitors, e.g., velpatasvir, pibrentasvir, elbasvir |
| NS5B | RNA-dependent RNA polymerase | NS5B inhibitor, e.g., sofosbuvir |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dietz, C.; Maasoumy, B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice. Viruses 2022, 14, 1325. https://doi.org/10.3390/v14061325
Dietz C, Maasoumy B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice. Viruses. 2022; 14(6):1325. https://doi.org/10.3390/v14061325
Chicago/Turabian StyleDietz, Christopher, and Benjamin Maasoumy. 2022. "Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice" Viruses 14, no. 6: 1325. https://doi.org/10.3390/v14061325
APA StyleDietz, C., & Maasoumy, B. (2022). Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice. Viruses, 14(6), 1325. https://doi.org/10.3390/v14061325