
SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Plasmids, and Reagents
2.2. Pseudovirus Production and Characterization
2.3. Infectivity Assay
2.4. Data Processing and Statistical Analysis
3. Results
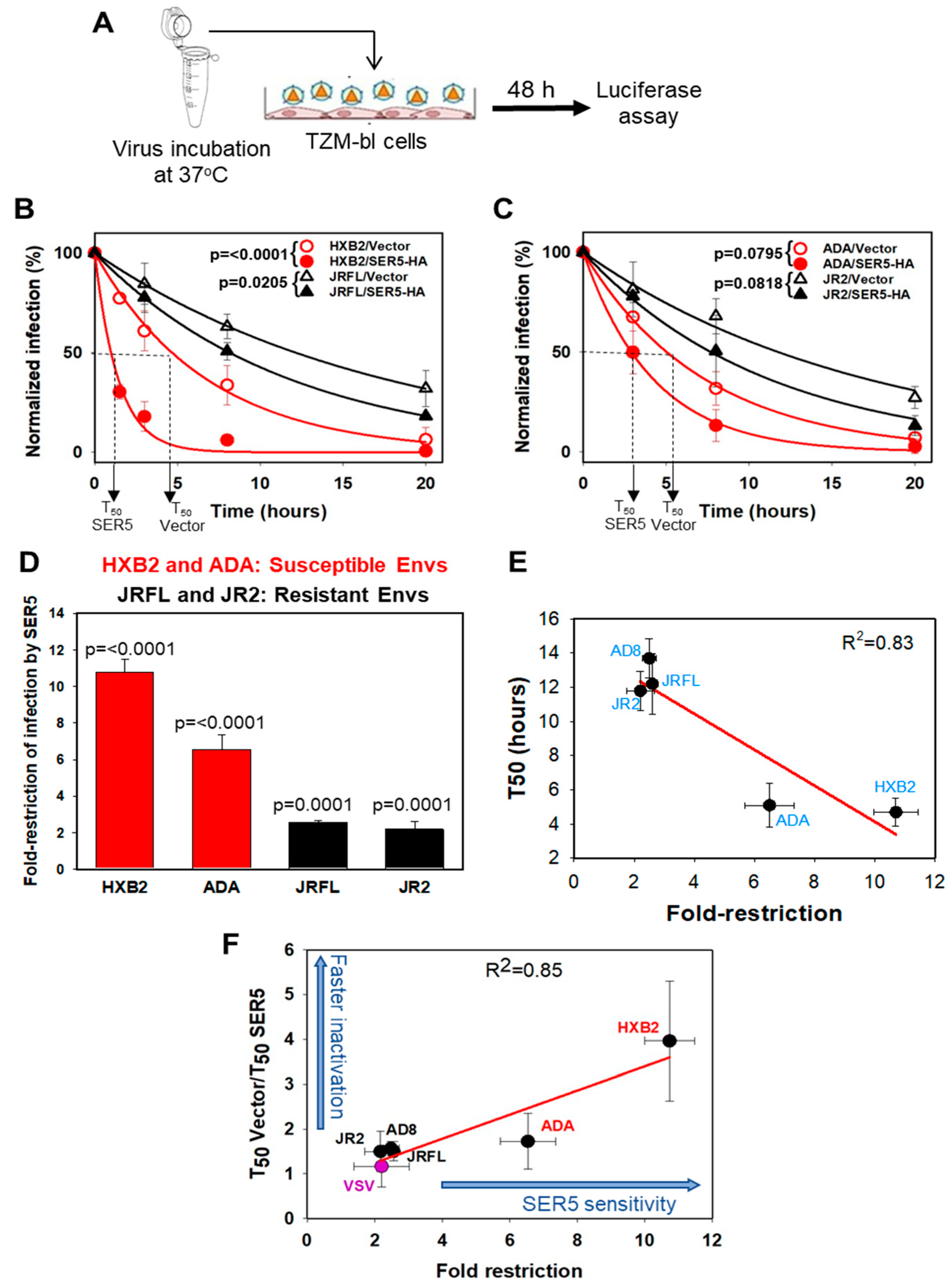
3.1. HIV-1 Sensitivity to SER5 Restriction Correlates with the Rate of Spontaneous Loss of Infectivity
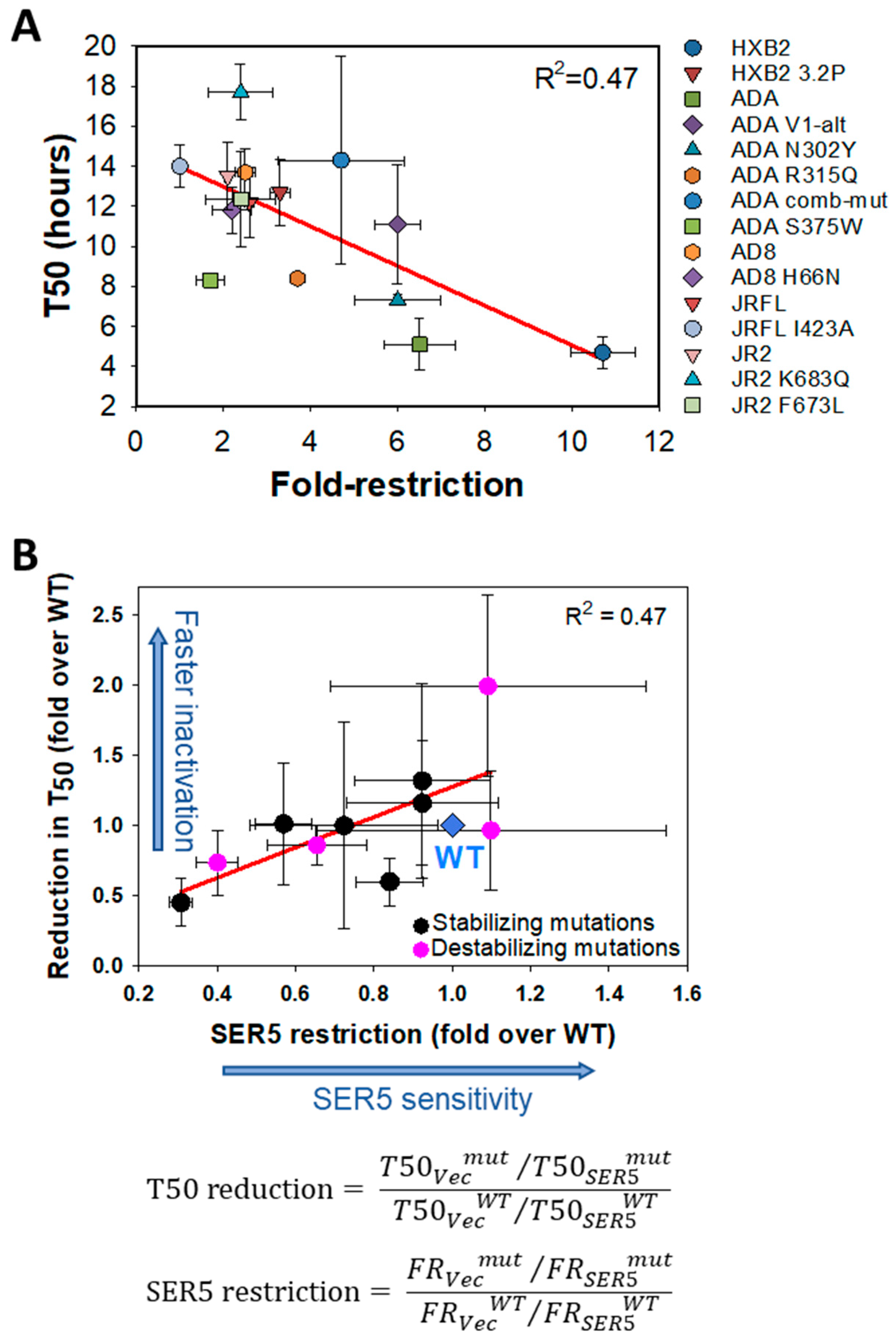
3.2. Mutations Disfavoring the Closed Conformation of Env Do Not Generally Sensitize HIV-1 to SER5 Restriction
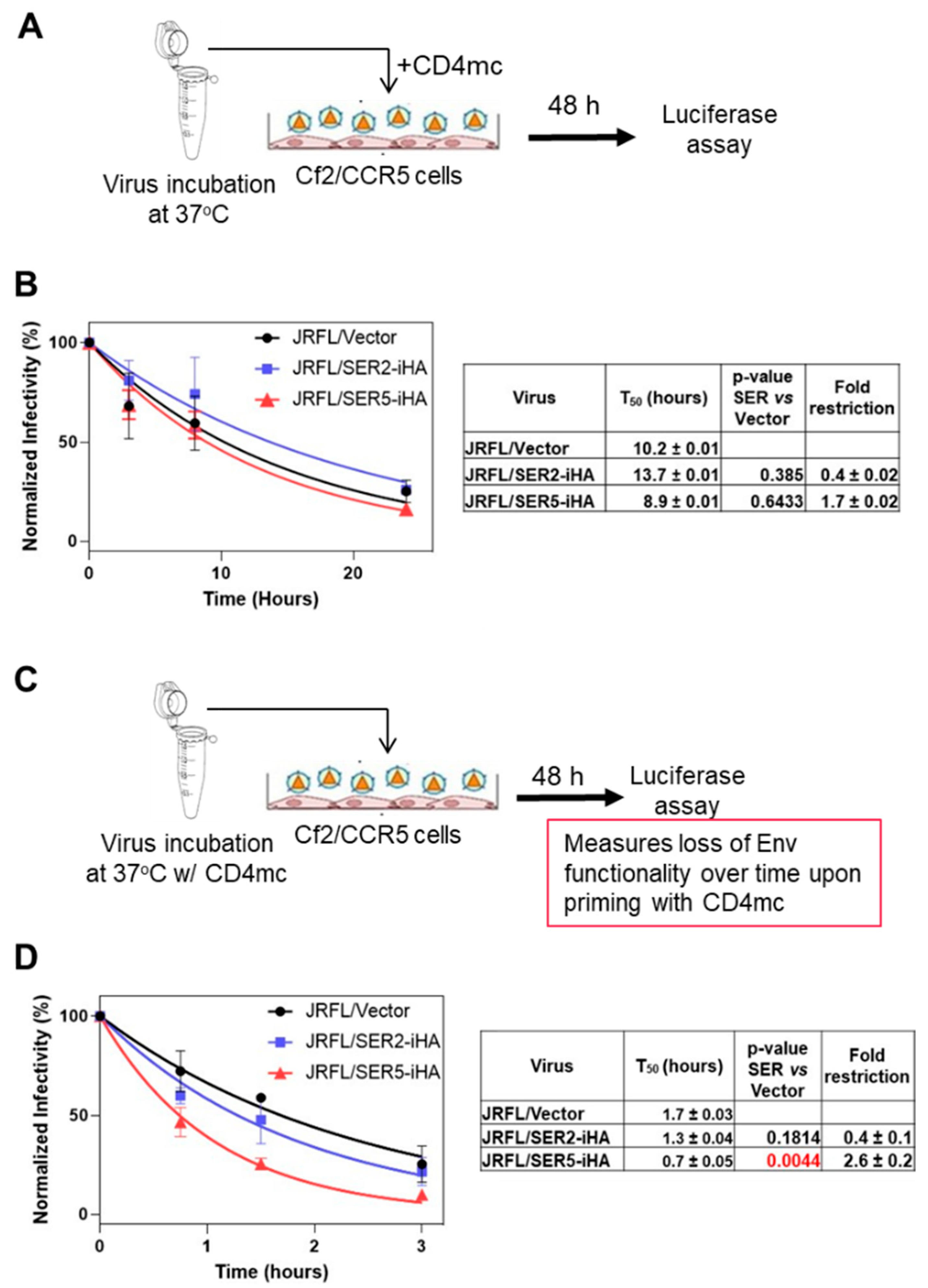
3.3. CD4-Bound Env Conformation Is More Sensitive to SER5 Restriction
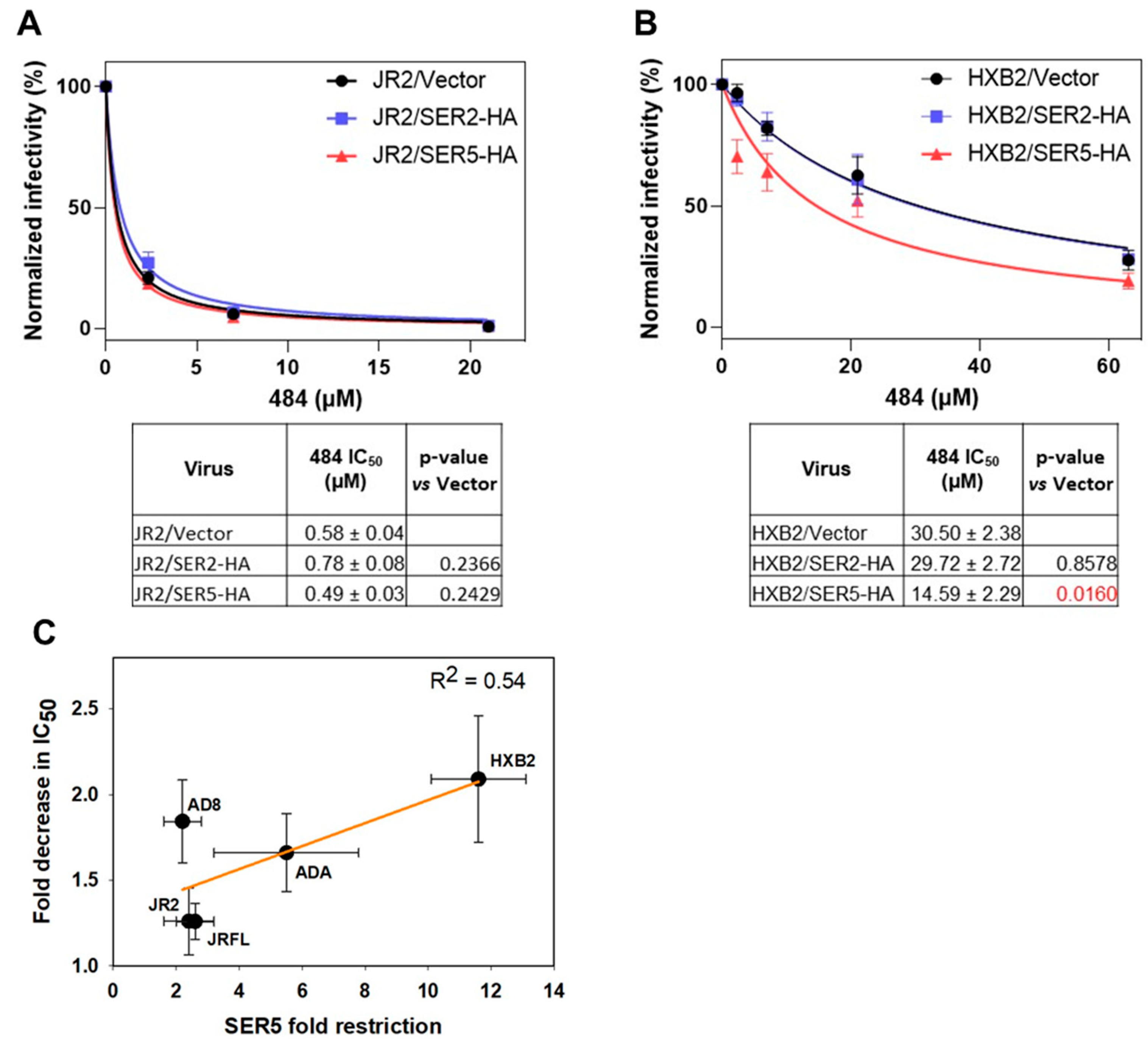
3.4. SER5 Sensitizes Env to Small Molecule Inhibitor Targeting the Closed Env
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Sood, C.; Marin, M.; Aaron, J.; Gratton, E.; Salaita, K.; Melikyan, G.B. Super-Resolution Fluorescence Imaging Reveals That Serine Incorporator Protein 5 Inhibits Human Immunodeficiency Virus Fusion by Disrupting Envelope Glycoprotein Clusters. ACS Nano 2020, 14, 10929–10943. [Google Scholar] [CrossRef]
- Schulte, B.; Selyutina, A.; Opp, S.; Herschhorn, A.; Sodroski, J.G.; Pizzato, M.; Diaz-Griffero, F. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 2018, 515, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Li, S.; Li, R.; Chai, Q.; Zhang, L.; Wang, B.; Yu, C.; Zheng, Y.H. The retroviral accessory proteins S2, Nef, and glycoMA use similar mechanisms for antagonizing the host restriction factor SERINC5. J. Biol. Chem. 2019, 294, 7013–7024. [Google Scholar] [CrossRef]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ahmad, I.; Shi, J.; Wang, B.; Yu, C.; Zhang, L.; Zheng, Y.H. Murine Leukemia Virus Glycosylated Gag Reduces Murine SERINC5 Protein Expression at Steady-State Levels via the Endosome/Lysosome Pathway to Counteract SERINC5 Antiretroviral Activity. J. Virol. 2019, 93, e01651-18. [Google Scholar] [CrossRef] [Green Version]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef]
- Trautz, B.; Wiedemann, H.; Luchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Krausslich, H.G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Biol. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [Green Version]
- Pye, V.E.; Rosa, A.; Bertelli, C.; Struwe, W.B.; Maslen, S.L.; Corey, R.; Liko, I.; Hassall, M.; Mattiuzzo, G.; Ballandras-Colas, A.; et al. A bipartite structural organization defines the SERINC family of HIV-1 restriction factors. Nat. Struct. Mol. Biol. 2020, 27, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.E.; Kiessling, V.; Pornillos, O.; White, J.M.; Ganser-Pornillos, B.K.; Tamm, L.K. HIV-cell membrane fusion intermediates are restricted by Serincs as revealed by cryo-electron and TIRF microscopy. J. Biol. Chem. 2020, 295, 15183–15195. [Google Scholar] [CrossRef] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Gottlinger, H. HIV-1 Nef responsiveness is determined by Env variable regions involved in trimer association and correlates with neutralization sensitivity. Cell Rep. 2013, 5, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. eLife 2018, 7, e34271. [Google Scholar] [CrossRef]
- Munro, J.B.; Nath, A.; Farber, M.; Datta, S.A.; Rein, A.; Rhoades, E.; Mothes, W. A conformational transition observed in single HIV-1 Gag molecules during in vitro assembly of virus-like particles. J. Virol. 2014, 88, 3577–3585. [Google Scholar] [CrossRef] [Green Version]
- Bowder, D.; Hollingsead, H.; Durst, K.; Hu, D.; Wei, W.; Wiggins, J.; Medjahed, H.; Finzi, A.; Sodroski, J.; Xiang, S.H. Contribution of the gp120 V3 loop to envelope glycoprotein trimer stability in primate immunodeficiency viruses. Virology 2018, 521, 158–168. [Google Scholar] [CrossRef]
- Rusert, P.; Krarup, A.; Magnus, C.; Brandenberg, O.F.; Weber, J.; Ehlert, A.K.; Regoes, R.R.; Gunthard, H.F.; Trkola, A. Interaction of the gp120 V1V2 loop with a neighboring gp120 unit shields the HIV envelope trimer against cross-neutralizing antibodies. J. Exp. Med. 2011, 208, 1419–1433. [Google Scholar] [CrossRef] [Green Version]
- Lyumkis, D.; Julien, J.P.; de Val, N.; Cupo, A.; Potter, C.S.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; Carragher, B.; et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shi, J.; Qiu, X.; Chai, Q.; Frabutt, D.A.; Schwartz, R.C.; Zheng, Y.H. CD4 Expression and Env Conformation Are Critical for HIV-1 Restriction by SERINC5. J. Virol. 2019, 93, e00544-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staropoli, I.; Dufloo, J.; Ducher, A.; Commere, P.H.; Sartori-Rupp, A.; Novault, S.; Bruel, T.; Lorin, V.; Mouquet, H.; Schwartz, O.; et al. Flow Cytometry Analysis of HIV-1 Env Conformations at the Surface of Infected Cells and Virions: Role of Nef, CD4, and SERINC5. J. Virol. 2020, 94, e01783-19. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, A.; Aiken, C. SERINC5 Inhibits HIV-1 Infectivity by Altering the Conformation of gp120 on HIV-1 Particles. J. Virol. 2020, 94, e00594-20. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Herschhorn, A.; Gu, C.; Espy, N.; Richard, J.; Finzi, A.; Sodroski, J.G. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat. Chem. Biol. 2014, 10, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzabekov, T.; Bannert, N.; Farzan, M.; Hofmann, W.; Kolchinsky, P.; Wu, L.; Wyatt, R.; Sodroski, J. Enhanced expression, native purification, and characterization of CCR5, a principal HIV-1 coreceptor. J. Biol. Chem. 1999, 274, 28745–28750. [Google Scholar] [CrossRef] [Green Version]
- Leaman, D.P.; Zwick, M.B. Increased functional stability and homogeneity of viral envelope spikes through directed evolution. PLoS Pathog. 2013, 9, e1003184. [Google Scholar] [CrossRef] [Green Version]
- Gift, S.K.; Leaman, D.P.; Zhang, L.; Kim, A.S.; Zwick, M.B. Functional Stability of HIV-1 Envelope Trimer Affects Accessibility to Broadly Neutralizing Antibodies at Its Apex. J. Virol. 2017, 91, e01216-17. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.S.; Leaman, D.P.; Zwick, M.B. Antibody to gp41 MPER alters functional properties of HIV-1 Env without complete neutralization. PLoS Pathog. 2014, 10, e1004271. [Google Scholar] [CrossRef]
- Xiang, S.H.; Kwong, P.D.; Gupta, R.; Rizzuto, C.D.; Casper, D.J.; Wyatt, R.; Wang, L.; Hendrickson, W.A.; Doyle, M.L.; Sodroski, J. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 2002, 76, 9888–9899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haim, H.; Strack, B.; Kassa, A.; Madani, N.; Wang, L.; Courter, J.R.; Princiotto, A.; McGee, K.; Pacheco, B.; Seaman, M.S.; et al. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog. 2011, 7, e1002101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Z.; Cayabyab, M.; Sodroski, J. Envelope glycoprotein determinants of neutralization resistance in a simian-human immunodeficiency virus (SHIV-HXBc2P 3.2) derived by passage in monkeys. J. Virol. 2001, 75, 4208–4218. [Google Scholar] [CrossRef] [Green Version]
- Herschhorn, A.; Gu, C.; Moraca, F.; Ma, X.; Farrell, M.; Smith, A.B., III; Pancera, M.; Kwong, P.D.; Schon, A.; Freire, E.; et al. The beta20-beta21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 2017, 8, 1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melillo, B.; Liang, S.; Park, J.; Schon, A.; Courter, J.R.; LaLonde, J.M.; Wendler, D.J.; Princiotto, A.M.; Seaman, M.S.; Freire, E.; et al. Small-Molecule CD4-Mimics: Structure-Based Optimization of HIV-1 Entry Inhibition. ACS Med. Chem. Lett. 2016, 7, 330–334. [Google Scholar] [CrossRef] [Green Version]
- Hammonds, J.; Chen, X.; Zhang, X.; Lee, F.; Spearman, P. Advances in methods for the production, purification, and characterization of HIV-1 Gag-Env pseudovirion vaccines. Vaccine 2007, 25, 8036–8048. [Google Scholar] [CrossRef]
- Agrawal, N.; Leaman, D.P.; Rowcliffe, E.; Kinkead, H.; Nohria, R.; Akagi, J.; Bauer, K.; Du, S.X.; Whalen, R.G.; Burton, D.R.; et al. Functional stability of unliganded envelope glycoprotein spikes among isolates of human immunodeficiency virus type 1 (HIV-1). PLoS ONE 2011, 6, e21339. [Google Scholar] [CrossRef] [Green Version]
- Kassa, A.; Finzi, A.; Pancera, M.; Courter, J.R.; Smith, A.B., III; Sodroski, J. Identification of a human immunodeficiency virus type 1 envelope glycoprotein variant resistant to cold inactivation. J. Virol. 2009, 83, 4476–4488. [Google Scholar] [CrossRef] [Green Version]
- Madani, N.; Princiotto, A.M.; Mach, L.; Ding, S.; Prevost, J.; Richard, J.; Hora, B.; Sutherland, L.; Zhao, C.A.; Conn, B.P.; et al. A CD4-mimetic compound enhances vaccine efficacy against stringent immunodeficiency virus challenge. Nat. Commun. 2018, 9, 2363. [Google Scholar] [CrossRef]
- Madani, N.; Princiotto, A.M.; Easterhoff, D.; Bradley, T.; Luo, K.; Williams, W.B.; Liao, H.X.; Moody, M.A.; Phad, G.E.; Vazquez Bernat, N.; et al. Antibodies Elicited by Multiple Envelope Glycoprotein Immunogens in Primates Neutralize Primary Human Immunodeficiency Viruses (HIV-1) Sensitized by CD4-Mimetic Compounds. J. Virol. 2016, 90, 5031–5046. [Google Scholar] [CrossRef] [Green Version]
- Beitari, S.; Pan, Q.; Finzi, A.; Liang, C. Differential Pressures of SERINC5 and IFITM3 on HIV-1 Envelope Glycoprotein over the Course of HIV-1 Infection. J. Virol. 2020, 94, e00514-20. [Google Scholar] [CrossRef] [PubMed]
- Haim, H.; Si, Z.; Madani, N.; Wang, L.; Courter, J.R.; Princiotto, A.; Kassa, A.; DeGrace, M.; McGee-Estrada, K.; Mefford, M.; et al. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 2009, 5, e1000360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattentau, Q.J.; Moore, J.P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415. [Google Scholar] [CrossRef]
- Diehl, W.E.; Guney, M.H.; Vanzo, T.; Kyawe, P.P.; White, J.M.; Pizzato, M.; Luban, J. Influence of Different Glycoproteins and of the Virion Core on SERINC5 Antiviral Activity. Viruses 2021, 13, 1279. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Garai, J.A.; Arboleya, A.; Otaegi, S.; Chojnacki, J.; Casas, J.; Fabrias, G.; Contreras, F.X.; Krausslich, H.G.; Lorizate, M. Cholesterol in the Viral Membrane is a Molecular Switch Governing HIV-1 Env Clustering. Adv. Sci. 2021, 8, 2003468. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirschman, J.; Marin, M.; Chen, Y.-C.; Chen, J.; Herschhorn, A.; Smith, A.B., III; Melikyan, G.B. SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env. Viruses 2022, 14, 1388. https://doi.org/10.3390/v14071388
Kirschman J, Marin M, Chen Y-C, Chen J, Herschhorn A, Smith AB III, Melikyan GB. SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env. Viruses. 2022; 14(7):1388. https://doi.org/10.3390/v14071388
Chicago/Turabian StyleKirschman, Junghwa, Mariana Marin, Yen-Cheng Chen, Junhua Chen, Alon Herschhorn, Amos B. Smith, III, and Gregory B. Melikyan. 2022. "SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env" Viruses 14, no. 7: 1388. https://doi.org/10.3390/v14071388
APA StyleKirschman, J., Marin, M., Chen, Y. -C., Chen, J., Herschhorn, A., Smith, A. B., III, & Melikyan, G. B. (2022). SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env. Viruses, 14(7), 1388. https://doi.org/10.3390/v14071388