
Evaluation of the HIV-1 Polymerase Gene Sequence Diversity for Prediction of Recent HIV-1 Infections Using Shannon Entropy Analysis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Nucleic Acid Extraction and Amplification of HIV-1 pol
2.3. Sanger Sequencing and Sequence Analysis
2.4. Shannon Entropy Analysis for Measuring Diversity
2.5. Reference Sequence Data Sources
2.6. Statistical Analysis
3. Results
3.1. Demographics
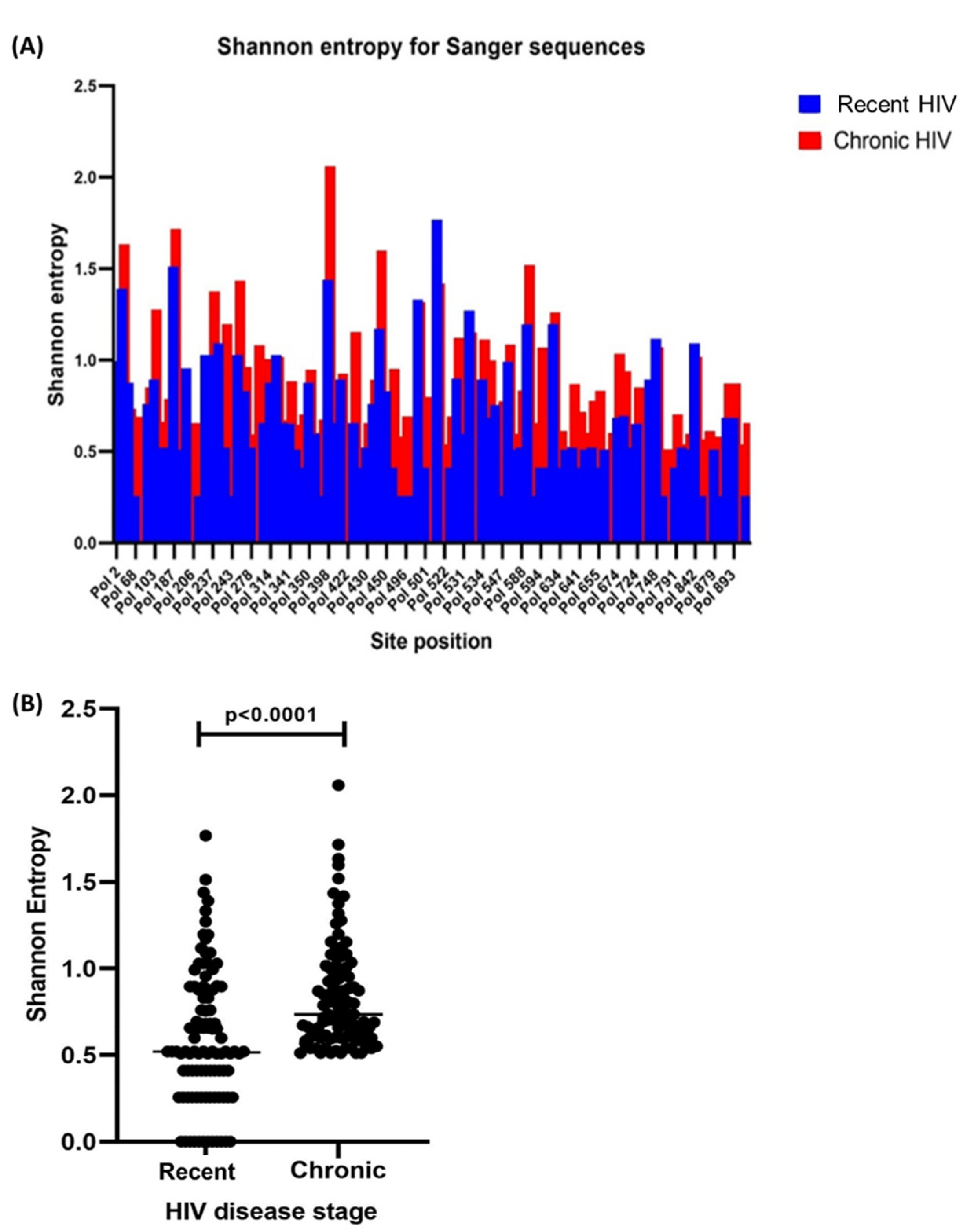
3.2. Sequencing and Complete HIV-1 pol Shannon Entropy
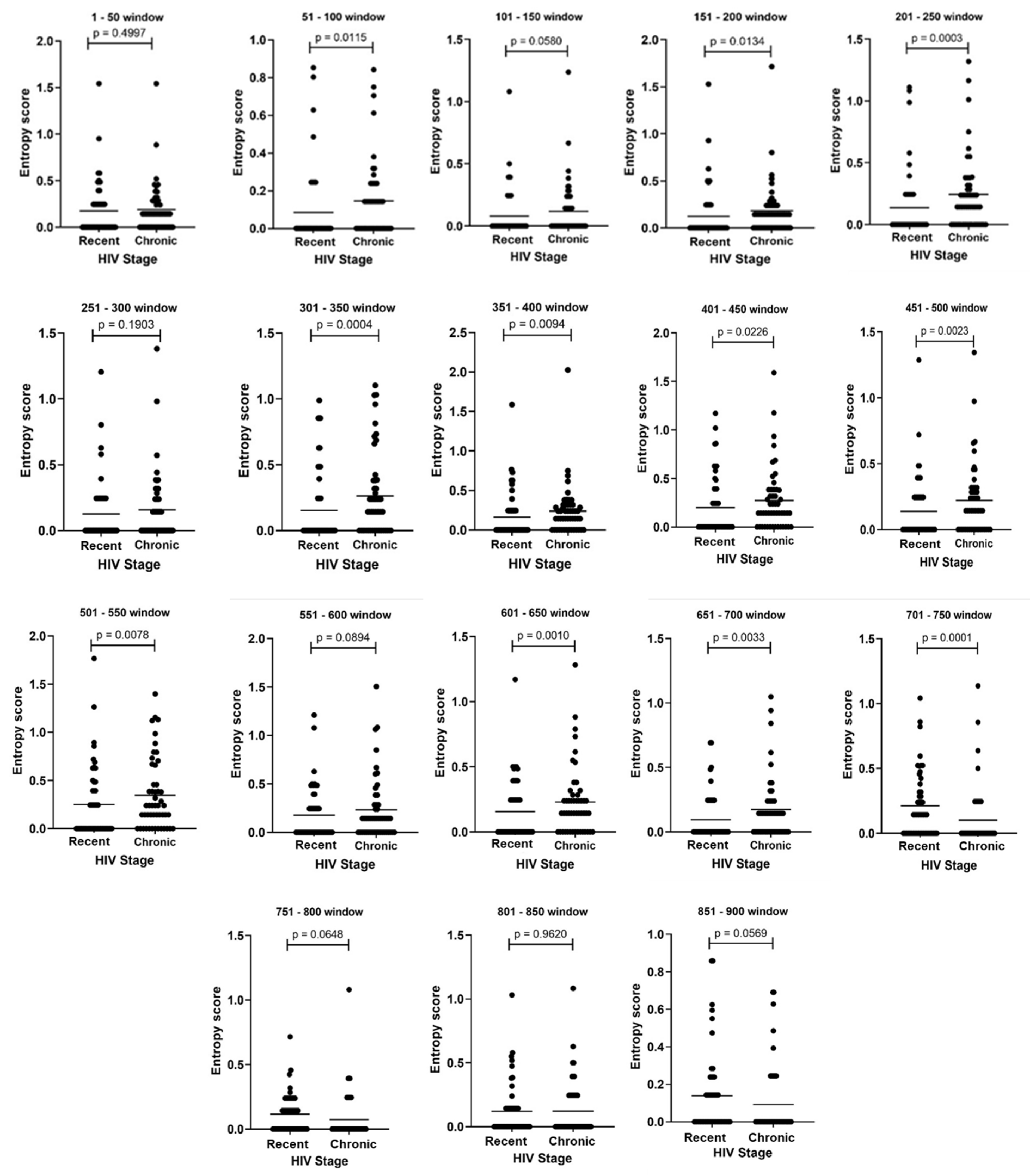
3.3. Amino Acid Diversity within HIV-1 pol Sliding Windows
3.4. A pol-Based Molecular Strategy to Detect Recent HIV-1 Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| AIDS | Acquired immunodeficiency syndrome |
| ART | Antiretroviral therapy |
| ARV | Antiretroviral |
| CD4 | Cluster of differentiation 4 |
| COVID-19 | Coronavirus disease of 2019 |
| CTL | Cytotoxic T-lymphocyte |
| dN-dS | Nonsynonymous to synonymous ratio |
| Env (env) | Envelope |
| FRR | False recency rate |
| Gag (gag) | Group-specific antigen |
| HIV-1 | Human immunodeficiency virus type 1 |
| IgG | Immunoglobulin G |
| IN | Integrase |
| IQR | Interquartile range |
| LAg | Limiting antigen avidity |
| LANL | Los Alamos National Laboratory |
| MAA | Multi-assay algorithm |
| NAAT | Nucleic acid amplification test |
| OD-n | Normalised optical density |
| ORF | Open reading frame |
| Pol (pol) | Polymerase |
| RT | Reverse transcriptase |
| RT-PCR | Reverse transcription polymerase chain reaction |
| VL | Viral load |
References
- Yusuf, T.T.; Benyah, F. Optimal strategy for controlling the spread of HIV/AIDS disease: A case study of South Africa. J. Biol. Dyn. 2012, 6, 475–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killian, M.S.; Levy, J.A. HIV/AIDS: 30 years of progress and future challenges. Eur. J. Immunol. 2011, 41, 3401–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Laboratory Methods for Diagnosis of HIV Infection in Infants and Children. In WHO Recommendations on the Diagnosis of HIV Infection in Infants and Children; World Health Organization Press: Geneva, Switzerland, 2010; pp. 509–635. Available online: https://www.ncbi.nlm.nih.gov/books/NBK138552/ (accessed on 12 January 2022).
- Meintjes, G.; Moorhouse, M.A.; Carmona, S.; Davies, N.; Dlamini, S.; Van Vuuren, C.; Manzini, T.; Mathe, M.; Moosa, Y.; Nash, J.; et al. Adult antiretroviral therapy guidelines 2017. S. Afr. J. HIV Med. 2017, 18, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoner, M.C.D.; Westreich, D.; Ahern, J.; Edwards, J.; Gómez-Olivé, F.X.; Tollman, S.M.; Lippman, S.; Kahn, K.; Pettifor, A. Modeling Combination Interventions to Prevent Human Immunodeficiency Virus in Adolescent Girls and Young Women in South Africa (HIV Prevention Trials Network 068). Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 73, e1911–e1918. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global 2020 HIV/AIDS Statistics. 2021. Available online: https://www.unaids.org/sites/default/files/media_asset/JC3032_AIDS_Data_book_2021_En.pdf (accessed on 3 February 2022).
- Jewell, B.L.; Mudimu, E.; Stover, J.; Ten Brink, D.; Phillips, A.N.; Smith, J.A.; Martin-Hughes, R.; Teng, Y.; Glaubius, R.; Mahiane, S.G.; et al. Potential effects of disruption to HIV programmes in sub-Saharan Africa caused by COVID-19: Results from multiple mathematical models. Lancet HIV 2020, 7, e629–e640. [Google Scholar] [CrossRef]
- Dorward, J.; Khubone, T.; Gate, K.; Ngobese, H.; Sookrajh, Y.; Mkhize, S.; Jeewa, A.; Bottomley, C.; Lewis, L.; Baisley, K.; et al. The impact of the COVID-19 lockdown on HIV care in 65 South African primary care clinics: An interrupted time series analysis. Lancet HIV 2021, 8, e158–e165. [Google Scholar] [CrossRef]
- Mastro, T.D.; Kim, A.A.; Hallett, T.; Rehle, T.; Welte, A.; Laeyendecker, O.; Oluoch, T.; Garcia-Calleja, J.M. Estimating HIV Incidence in Populations Using Tests for Recent Infection: Issues, Challenges and the Way Forward. J. HIV AIDS Surveill. Epidemiol. 2010, 2, 1–14. [Google Scholar]
- Morrison, D.; Laeyendecker, O.; Brookmeyer, R. Cross-sectional HIV incidence estimation in an evolving epidemic. Stat. Med. 2019, 38, 3614–3627. [Google Scholar] [CrossRef]
- Fellows, I.E.; Shiraishi, R.W.; Cherutich, P.; Achia, T.; Young, P.W.; Kim, A.A. A new method for estimating HIV incidence from a single cross-sectional survey. PLoS ONE 2020, 15, e0237221. [Google Scholar] [CrossRef]
- O’Cofaigh, E.; Lewthwaite, P. Natural history of HIV and AIDS. Medicine 2013, 41, 411–416. [Google Scholar] [CrossRef]
- Tiemessen, C.T.; Martinson, N. Elite controllers: Understanding natural suppressive control of HIV-1 infection. CMEJ 2012, 30, 282–285. [Google Scholar]
- Konikoff, J.; Brookmeyer, R.; Longosz, A.F.; Cousins, M.M.; Celum, C.; Buchbinder, S.P.; Seage, G.R.; Kirk, G.D.; Moore, R.D.; Mehta, S.H.; et al. Performance of a Limiting-Antigen Avidity Enzyme Immunoassay for Cross-Sectional Estimation of HIV Incidence in the United States. PLoS ONE 2013, 8, e82772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laeyendecker, O.; Gray, R.H.; Grabowski, M.K.; Reynolds, S.; Ndyanabo, A.; Ssekasanvu, J.; Fernandez, R.E.; Wawer, M.J.; Serwadda, D.; Quinn, T.C. Validation of the Limiting Antigen Avidity Assay to Estimate Level and Trends in HIV Incidence in an A/D Epidemic in Rakai, Uganda. AIDS Res. Hum. Retrovir. 2019, 35, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Moyo, S.; Wilkinson, E.; Novitsky, V.; Vandormael, A.; Gaseitsiwe, S.; Essex, M.; Engelbrecht, S.; de Oliveira, T. Identifying Recent HIV Infections: From Serological Assays to Genomics. Viruses 2015, 7, 5508–5524. [Google Scholar] [CrossRef] [Green Version]
- Sedia, Biosciences, Corporation. Sedia HIV-1 LAg-avidity EIA. Portland (2013). Available online: https://www.sediabio.com/wp-content/uploads/2021/05/LN-6039-09PackageInsertLAgAvidityEIA.pdf (accessed on 25 May 2022).
- Mayaphi, S.H.; Martin, D.J.; Quinn, T.C.; Laeyendecker, O.; Olorunju, S.A.S.; Tintinger, G.R.; Stoltz, A.C. Detection of Acute and Early HIV-1 Infections in an HIV Hyper-Endemic Area with Limited Resources. PLoS ONE 2016, 11, e0164943. [Google Scholar] [CrossRef] [Green Version]
- Nkone, P.; Loubser, S.; Quinn, T.C.; Redd, A.D.; Ismail, A.; Tiemessen, C.T.; Mayaphi, S.H. Deep sequencing of the HIV-1 polymerase gene for characterisation of cytotoxic T-lymphocyte epitopes during early and chronic disease stages. Virol. J. 2022, 19, 56. [Google Scholar] [CrossRef]
- Mayaphi, S.H. Detection and Characterisation of Primary (Acute and Early) HIV-1 Infections in an HIV Hyper-Endemic Area. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2018. Available online: https://repository.up.ac.za/handle/2263/69914 (accessed on 19 February 2022).
- Treurnicht, F.; Seoighe, C.; Martin, D.; Wood, N.; Abrahams, M.-R.; Rosa, D.D.A.; Bredell, H.; Woodman, Z.; Hide, W.; Mlisana, K.; et al. Adaptive changes in HIV-1 subtype C proteins during early infection are driven by changes in HLA-associated immune pressure. Virology 2010, 396, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Manak, M.; Sina, S.; Anekella, B.; Hewlett, I.; Sanders-Buell, E.; Ragupathy, V.; Kim, J.; Vermeulen, M.; Stramer, S.L.; Sabino, E.; et al. Pilot Studies for Development of an HIV Subtype Panel for Surveillance of Global Diversity. AIDS Res. Hum. Retrovir. 2012, 28, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Parrish, N.F.; Wilen, C.; Banks, L.B.; Iyer, S.S.; Pfaff, J.M.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Decker, J.M.; Parrish, E.H.; Berg, A.; et al. Transmitted/Founder and Chronic Subtype C HIV-1 Use CD4 and CCR5 Receptors with Equal Efficiency and Are Not Inhibited by Blocking the Integrin α4β7. PLoS Pathog. 2012, 8, e1002686. [Google Scholar] [CrossRef]
- Sanchez, A.M.; DeMarco, C.T.; Hora, B.; Keinonen, S.; Chen, Y.; Brinkley, C.; Stone, M.; Tobler, L.; Keating, S.; Schito, M.; et al. Development of a contemporary globally diverse HIV viral panel by the EQAPOL program. J. Immunol. Methods 2014, 409, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Matthews, P.C.; Adland, E.; Listgarten, J.; Leslie, A.; Mkhwanazi, N.; Carlson, J.M.; Harndahl, M.; Stryhn, A.; Payne, R.P.; Ogwu, A.; et al. HLA-A*7401–Mediated Control of HIV Viremia Is Independent of Its Linkage Disequilibrium with HLA-B*5703. J. Immunol. 2011, 186, 5675–5686. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.A.; Montano, M.A.; McLane, M.F.; Renjifo, B.; Vannberg, F.; Foley, B.T.; Ndung’U, T.P.; Rahman, M.; Makhema, M.J.; Marlink, R.; et al. Molecular Cloning and Phylogenetic Analysis of Human Immunodeficiency Virus Type 1 Subtype C: A Set of 23 Full-Length Clones from Botswana. J. Virol. 1999, 73, 4427–4432. [Google Scholar] [CrossRef] [Green Version]
- Gonese, E.; Kilmarx, P.H.; Van Schalkwyk, C.; Grebe, E.; Mutasa, K.; Ntozini, R.; Parekh, B.; Dobbs, T.; Pottinger, Y.D.; Masciotra, S.; et al. Evaluation of the Performance of Three Biomarker Assays for Recent HIV Infection Using a Well-Characterized HIV-1 Subtype C Incidence Cohort. AIDS Res. Hum. Retrovir. 2019, 35, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Laeyendecker, O.; Brookmeyer, R.; Oliver, A.E.; Mullis, C.E.; Eaton, K.P.; Mueller, A.C.; Jacobson, L.P.; Margolick, J.B.; Brown, J.; Rinaldo, C.R.; et al. Factors Associated with Incorrect Identification of Recent HIV Infection Using the BED Capture Immunoassay. AIDS Res. Hum. Retrovir. 2012, 28, 816–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragonnet-Cronin, M.; Aris-Brosou, S.; Joanisse, I.; Merks, H.; Vallée, D.; Caminiti, K.; Rekart, M.; Krajden, M.; Cook, D.; Kim, J.; et al. Genetic Diversity as a Marker for Timing Infection in HIV-Infected Patients: Evaluation of a 6-Month Window and Comparison With BED. J. Infect. Dis. 2012, 206, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Kouyos, R.D.; von Wyl, V.; Yerly, S.; Böni, J.; Rieder, P.; Joos, B.; Taffé, P.; Shah, C.; Bürgisser, P.; Klimkait, T.; et al. Ambiguous nucleotide calls from population-based sequencing of HIV-1 are a marker for viral diversity and the age of infection. Clin. Infect. Dis. 2011, 52, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adland, E.; Hill, M.; Lavandier, N.; Csala, A.; Edwards, A.; Chen, F.; Radkowski, M.; Kowalska, J.D.; Paraskevis, D.; Hatzakis, A.; et al. Differential Immunodominance Hierarchy of CD8 + T-Cell Responses in HLA-B*27:05- and -B*27:02-Mediated Control of HIV-1 Infection. J. Virol. 2018, 92, e01685-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; McNevin, J.P.; Rolland, M.; Zhao, H.; Deng, W.; Maenza, J.; Stevens, C.E.; Collier, A.C.; McElrath, M.J.; Mullins, J.I. Conserved HIV-1 Epitopes Continuously Elicit Subdominant Cytotoxic T-Lymphocyte Responses. J. Infect. Dis. 2009, 200, 1825–1833. [Google Scholar] [CrossRef]
- Wu, J.W.; Patterson-Lomba, O.; Novitsky, V.; Pagano, M. A Generalized Entropy Measure of Within-Host Viral Diversity for Identifying Recent HIV-1 Infections. Medicine 2015, 94, e1865. [Google Scholar] [CrossRef]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011, 11, 45–56. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- Parekh, B.S.; Kennedy, M.S.; Dobbs, T.; Pau, C.-P.; Byers, R.; Green, T.; Hu, D.J.; Vanichseni, S.; Young, N.L.; Choopanya, K.; et al. Quantitative Detection of Increasing HIV Type 1 Antibodies after Seroconversion: A Simple Assay for Detecting Recent HIV Infection and Estimating Incidence. AIDS Res. Hum. Retrovir. 2002, 18, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.S.; Duong, Y.T.; Le, L.-V.; Tuan, N.A.; Parekh, B.S.; Ha, H.T.T.; Pham, Q.D.; Cuc, C.T.T.; Dobbs, T.; Tram, T.H.; et al. Estimating False-Recent Classification for the Limiting-Antigen Avidity EIA and BED-Capture Enzyme Immunoassay in Vietnam: Implications for HIV-1 Incidence Estimates. AIDS Res. Hum. Retrovir. 2017, 33, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Santos-Hoevener, C.; Meixenberger, K.; Zimmermann, R.; Somogyi, S.; Fiedler, S.; Hofmann, A.; Bartmeyer, B.; Jansen, K.; Hamouda, O.; et al. Improved Testing of Recent HIV-1 Infections with the BioRad Avidity Assay Compared to the Limiting Antigen Avidity Assay and BED Capture Enzyme Immunoassay: Evaluation Using Reference Sample Panels from the German Seroconverter Cohort. PLoS ONE 2014, 9, e98038. [Google Scholar] [CrossRef] [Green Version]
- Nikolopoulos, G.K.; Katsoulidou, A.; Kantzanou, M.; Rokka, C.; Tsiara, C.; Sypsa, V.; Paraskevis, D.; Psichogiou, M.; Friedman, S.; Hatzakis, A. Evaluation of the limiting antigen avidity EIA (LAg) in people who inject drugs in Greece. Epidemiol. Infect. 2016, 145, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Onsongo, S.; Abidi, S.H.; Khamadi, S.; Shah, R.; Kageha, S.; Ojwang, P.; Ali, S.; Okinda, N. Prevalence of Transmitted Drug Resistance Mutations in HIV-1-Infected Drug-Naive Patients from Urban and Suburban Regions of Kenya. AIDS Res. Hum. Retrovir. 2016, 32, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Meixenberger, K.; Yousef, K.P.; Smith, M.R.; Somogyi, S.; Fiedler, S.; Bartmeyer, B.; Hamouda, O.; Bannert, N.; Von Kleist, M.; Kücherer, C. Molecular evolution of HIV-1 integrase during the 20 years prior to the first approval of integrase inhibitors. Virol. J. 2017, 14, 223. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
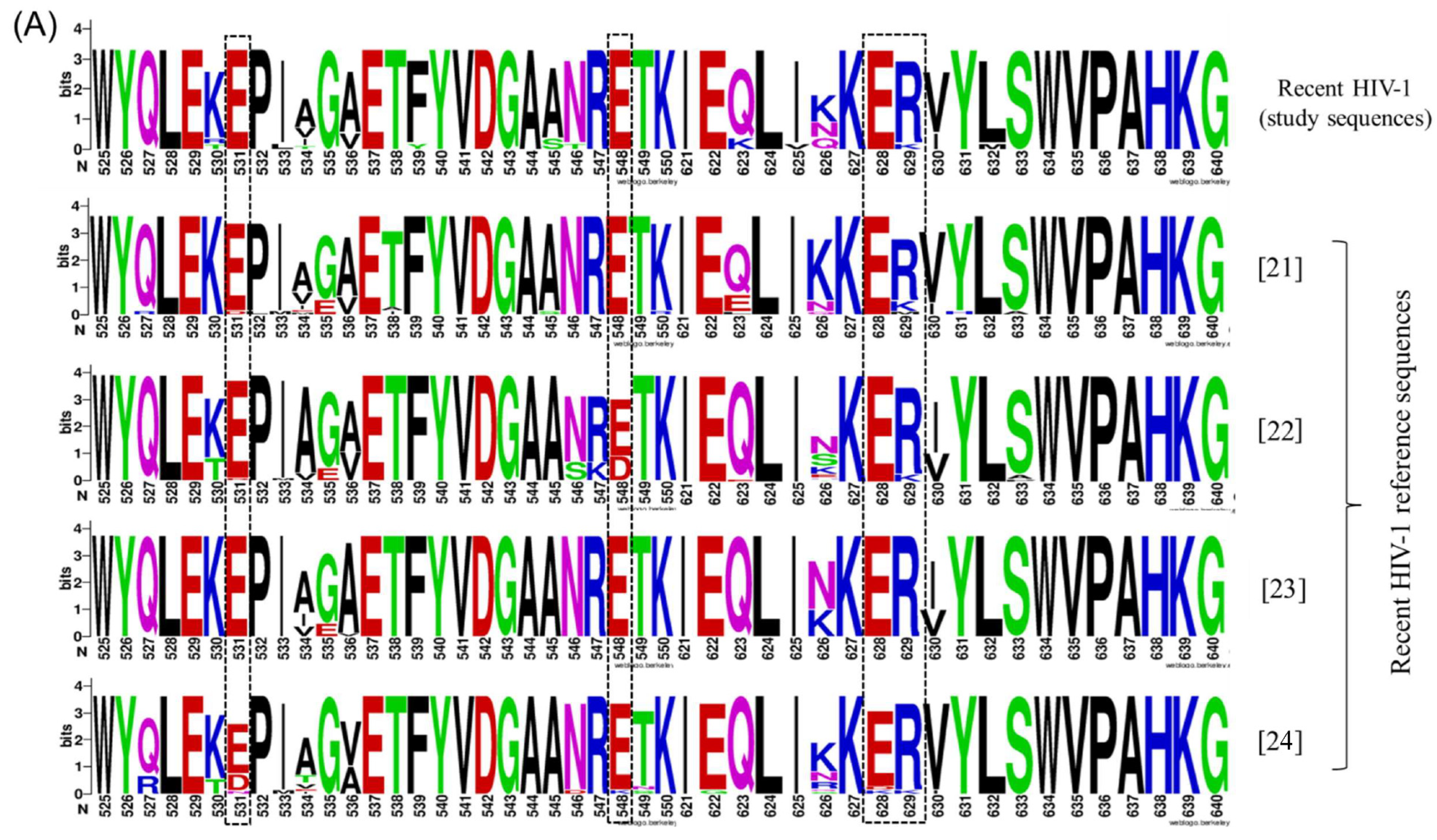{kind=link}
{kind=link}
| IT | ER | EL | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt ID | HIV Stage | Sex | Age (years) | HIV VL (copies/mL) | CD4 Count (cells/uL) | I234 | I241 | M456 | T499 | E531 | E548 | E628 | R629 | E670 | E684 | I690 | L704 | L733 |
| 261 | R | M | 33 | 386 | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 2504 | R | F | 24 | 287 | X | - | X | - | - | - | - | - | - | - | X | - | X | |
| 5041 | R | M | 23 | n/a | - | - | - | - | - | - | - | - | - | - | - | - | ||
| 6512 | R | F | 23 | 215 | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 6727 | R | F | 28 | 706 | X | - | - | - | - | - | - | - | - | - | - | - | - | |
| 6638 | R | F | 28 | 457 | X | - | - | - | - | - | - | - | - | - | - | - | - | |
| 6582 | R | F | 24 | 818 | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 9498 | R | F | 34 | 964 | - | - | - | X | - | - | - | - | - | X | - | - | - | |
| 9049 | R | F | 20 | n/a | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 8575 | R | F | 27 | 668 | - | - | X | X | - | - | - | - | - | X | - | - | - | |
| 8047 | R | M | 31 | n/a | X | - | - | - | - | - | - | - | - | - | X | - | - | |
| 7084 | R | F | 28 | 411 | - | - | X | - | - | - | - | - | X | - | - | - | - | |
| 6743 | R | F | 26 | 638 | - | - | - | - | - | - | - | - | - | - | - | - | X | |
| 6737 | R | F | 24 | n/a | X | - | - | - | - | - | - | - | - | - | - | - | X | |
| 639 | C | F | 30 | 392 | X | - | - | - | X | - | - | - | - | - | - | X | - | |
| 843 | C | F | 21 | 348 | - | X | X | - | - | - | - | X | - | - | - | - | - | |
| 1121 | C | F | 27 | 127 | - | - | - | - | - | X | - | - | X | - | - | - | - | |
| 1213 | C | F | 36 | n/a | - | - | - | - | - | X | - | - | - | - | - | - | - | |
| 1475 | C | F | 32 | 255 | - | - | X | X | - | X | X | - | - | - | X | - | - | |
| 2340 | C | F | 22 | n/a | - | - | - | - | - | - | - | X | - | X | X | - | - | |
| 2696 | C | F | 18 | n/a | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 3253 | C | F | 28 | n/a | - | - | - | - | X | - | - | X | X | X | X | - | - | |
| 3387 | C | F | 37 | n/a | - | - | X | - | - | X | - | - | - | - | - | - | - | |
| 3474 | C | F | 21 | 675 | - | - | X | - | - | - | - | - | X | - | - | - | - | |
| 9986 | C | F | 28 | 160 | - | X | X | - | - | - | - | - | - | - | X | - | - | |
| 3606 | C | F | 24 | 529 | - | - | - | - | - | - | - | - | - | - | - | X | - | |
| 3869 | C | F | 32 | n/a | - | - | X | - | - | - | - | - | - | - | X | - | - | |
| 3880 | C | F | 30 | 575 | - | - | - | - | - | - | - | - | X | - | - | X | - | |
| 3910 | C | F | 33 | n/a | - | - | - | - | X | - | X | - | - | - | - | X | - | |
| 3912 | C | F | 27 | 287 | - | - | - | - | - | - | - | X | X | - | - | - | - | |
| 3920 | C | F | 20 | 306 | - | - | X | - | - | - | - | X | - | - | - | - | - | |
| 4351 | C | F | 35 | n/a | - | X | X | - | X | - | - | - | - | - | - | - | - | |
| 4198 | C | F | 19 | n/a | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 5054 | C | F | 26 | 72 | - | - | X | - | - | - | - | - | - | - | - | - | - | |
| 6380 | C | F | 25 | 385 | - | X | - | - | - | - | - | - | - | - | - | X | - | |
| 6565 | C | F | 28 | 371 | - | - | - | - | - | - | - | - | - | X | - | - | - | |
| 6671 | C | F | 35 | n/a | - | - | - | X | - | X | - | - | - | - | - | - | - | |
| 6649 | C | F | 32 | 164 | - | - | X | X | - | - | - | - | - | - | X | - | - | |
| 6640 | C | F | 37 | 407 | - | - | - | - | - | - | X | - | - | - | X | X | - | |
| 6596 | C | F | 31 | 576 | X | - | - | - | X | - | - | X | - | - | X | - | - | |
| 9915 | C | F | 30 | 382 | - | - | - | X | - | - | - | - | - | X | - | - | - | |
| 9895 | C | F | 31 | 607 | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 9854 | C | F | 20 | n/a | - | - | X | - | - | - | - | - | X | - | - | X | - | |
| 7959 | C | F | 40 | 343 | - | - | X | X | - | - | X | - | X | X | - | - | - | |
| 6990 | C | F | 26 | 269 | - | - | - | X | - | - | - | - | - | - | - | - | - | |
| Amino Acid Site | CTL Epitope Mapped to | HIV-1 pol Position | Subtype Identified for |
|---|---|---|---|
| I241 | NETPGIRYQ | RT (137–145) | B |
| M456 | RMRTAHTNDVK | RT (356–366) | B, C |
| T499 | PIQKETWETW | RT (392–401) | B |
| E531 | EPIAGAETFY | RT (432–441) | C |
| E548 | RETKLGKAGY | RT (448–457) | -- |
| E628 | IKKEEVYLA | RT (526–534) | B |
| E629 | IKKEEVYLA | RT (526–534) | B |
| E670 | QEEHEKYHNSW | IN (9–19) | B, C |
| E684 | RAMASEFNL | IN (20–28) | -- |
| I690 | FNLPPIVAKEI | IN (26–36) | A |
| L704 | VASCDKCQL | IN (37–45) | C |
| IT | ER | EL | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study Sequences | n | I241 | M456 | T499 | E531 | E548 | E628 | R629 | E670 | E684 | I690 | L704 |
| Recent | 14 | 0 | 3 | 2 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 0 |
| Chronic | 30 | 4 | 12 | 6 | 5 | 5 | 4 | 6 | 7 | 5 | 8 | 7 |
| Recent reference sequences | ||||||||||||
| SA [21] | 21 | 2 | 5 | 1 | 1 | 0 | 0 | 4 | 5 | 3 | 5 | 3 |
| SA [22] | 29 | 2 | 6 | 16 | 1 | 8 | 0 | 2 | 2 | 3 | 8 | 1 |
| Malawi [23] | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 9 | 15 |
| India [24] | 21 | 1 | 3 | 2 | 6 | 1 | 2 | 1 | 3 | 21 | 8 | 2 |
| Chronic reference sequences | ||||||||||||
| SA [25] | 75 | 3 | 23 | 21 | 7 | 8 | 5 | 5 | 7 | 12 | 15 | 12 |
| Malawi [23] | 38 | 0 | 6 | 0 | 0 | 9 | 0 | 5 | 9 | 7 | 0 | 10 |
| Botswana [26] | 23 | 7 | 12 | 10 | 11 | 6 | 3 | 14 | 6 | 15 | 19 | 0 |
| Stage | n | ER | ERM | EL | ELM | IT |
|---|---|---|---|---|---|---|
| <6 months | 14 | 14 (100%) | 11 (78.6%) | 9 (64.3%) | 9 (64.3%) | 10 (71.4%) |
| >6 months | 31 | 15 (48.4%) | 9 (29.0%) | 11 (35.5%) | 6 (19.4%) | 15 (48.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nkone, P.; Loubser, S.; Quinn, T.C.; Redd, A.D.; Laeyendecker, O.; Tiemessen, C.T.; Mayaphi, S.H. Evaluation of the HIV-1 Polymerase Gene Sequence Diversity for Prediction of Recent HIV-1 Infections Using Shannon Entropy Analysis. Viruses 2022, 14, 1587. https://doi.org/10.3390/v14071587
Nkone P, Loubser S, Quinn TC, Redd AD, Laeyendecker O, Tiemessen CT, Mayaphi SH. Evaluation of the HIV-1 Polymerase Gene Sequence Diversity for Prediction of Recent HIV-1 Infections Using Shannon Entropy Analysis. Viruses. 2022; 14(7):1587. https://doi.org/10.3390/v14071587
Chicago/Turabian StyleNkone, Paballo, Shayne Loubser, Thomas C. Quinn, Andrew D. Redd, Oliver Laeyendecker, Caroline T. Tiemessen, and Simnikiwe H. Mayaphi. 2022. "Evaluation of the HIV-1 Polymerase Gene Sequence Diversity for Prediction of Recent HIV-1 Infections Using Shannon Entropy Analysis" Viruses 14, no. 7: 1587. https://doi.org/10.3390/v14071587
APA StyleNkone, P., Loubser, S., Quinn, T. C., Redd, A. D., Laeyendecker, O., Tiemessen, C. T., & Mayaphi, S. H. (2022). Evaluation of the HIV-1 Polymerase Gene Sequence Diversity for Prediction of Recent HIV-1 Infections Using Shannon Entropy Analysis. Viruses, 14(7), 1587. https://doi.org/10.3390/v14071587