
A Human and Rhesus Macaque Interferon-Stimulated Gene Screen Shows That Over-Expression of ARHGEF3/XPLN Inhibits Replication of Hepatitis C Virus and Other Flavivirids

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Molecular Biology
2.3. Generation of ISG Lentivirus Library
2.4. HCV Reagents
2.5. YFV and ZIKV Reporter Assays
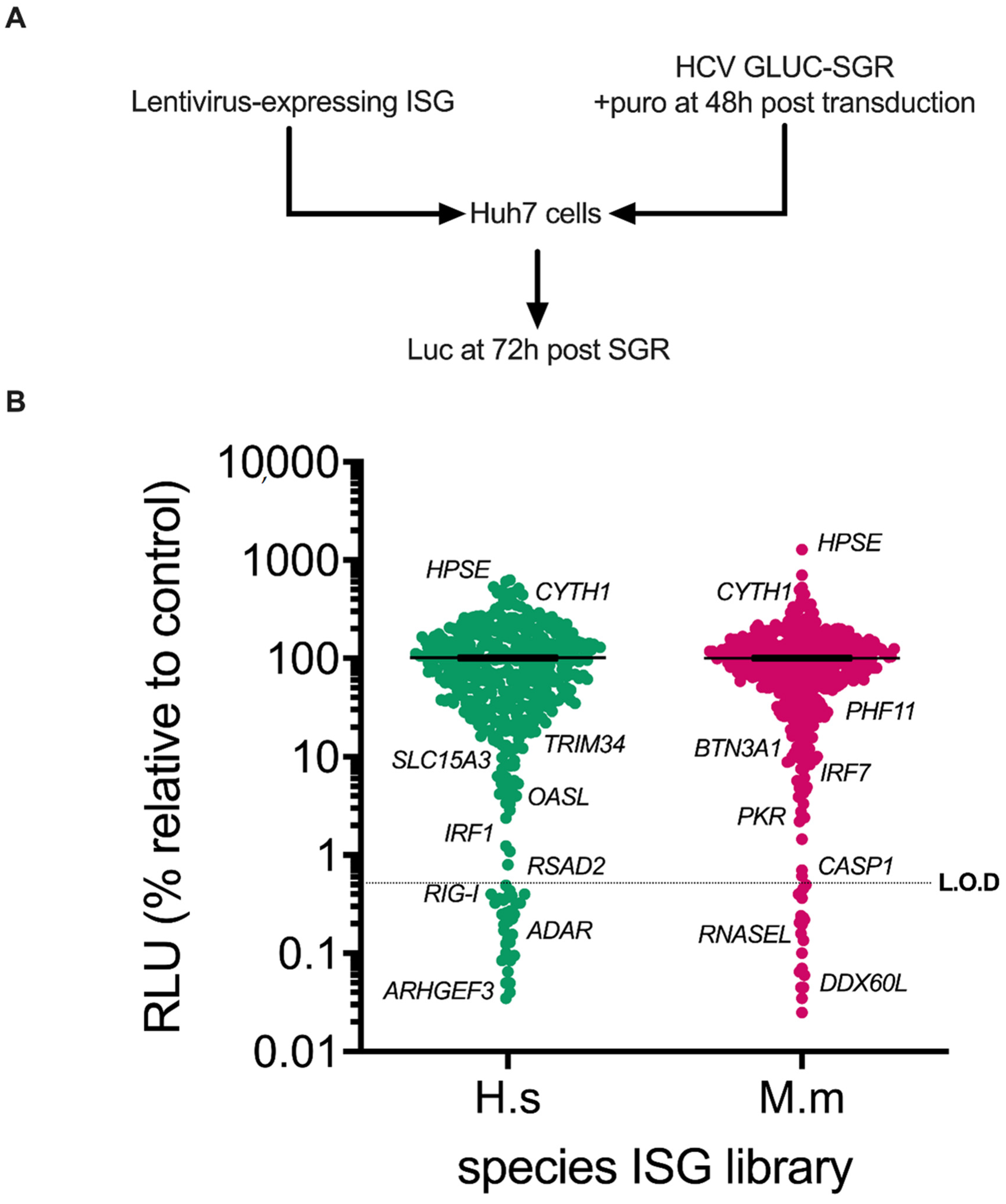
2.6. ISG Screening Protocol
2.7. Determination of Transduction Efficiency
2.8. RT-qPCR
2.9. Immunoblotting
2.10. Bioinformatic Analysis of ISG Expression in HCV-Infected Human Livers
2.11. Statistical Analyses and Reproducibility
3. Results
3.1. Screening of Human and Rhesus Macaque ISGs against HCV Sub-Genomic Replicon Replication
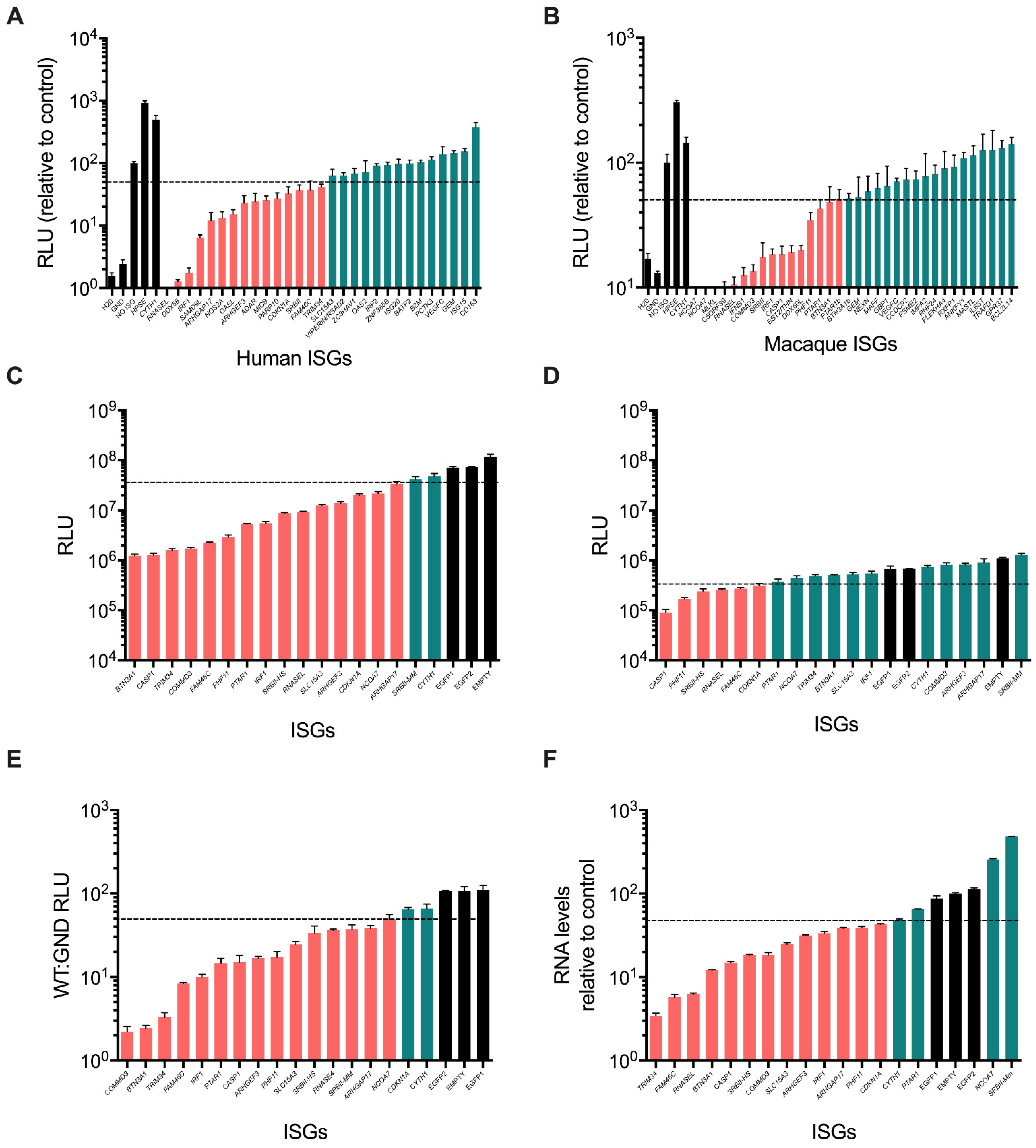
3.2. Validation of ISG Inhibitors of HCV SGR Replication
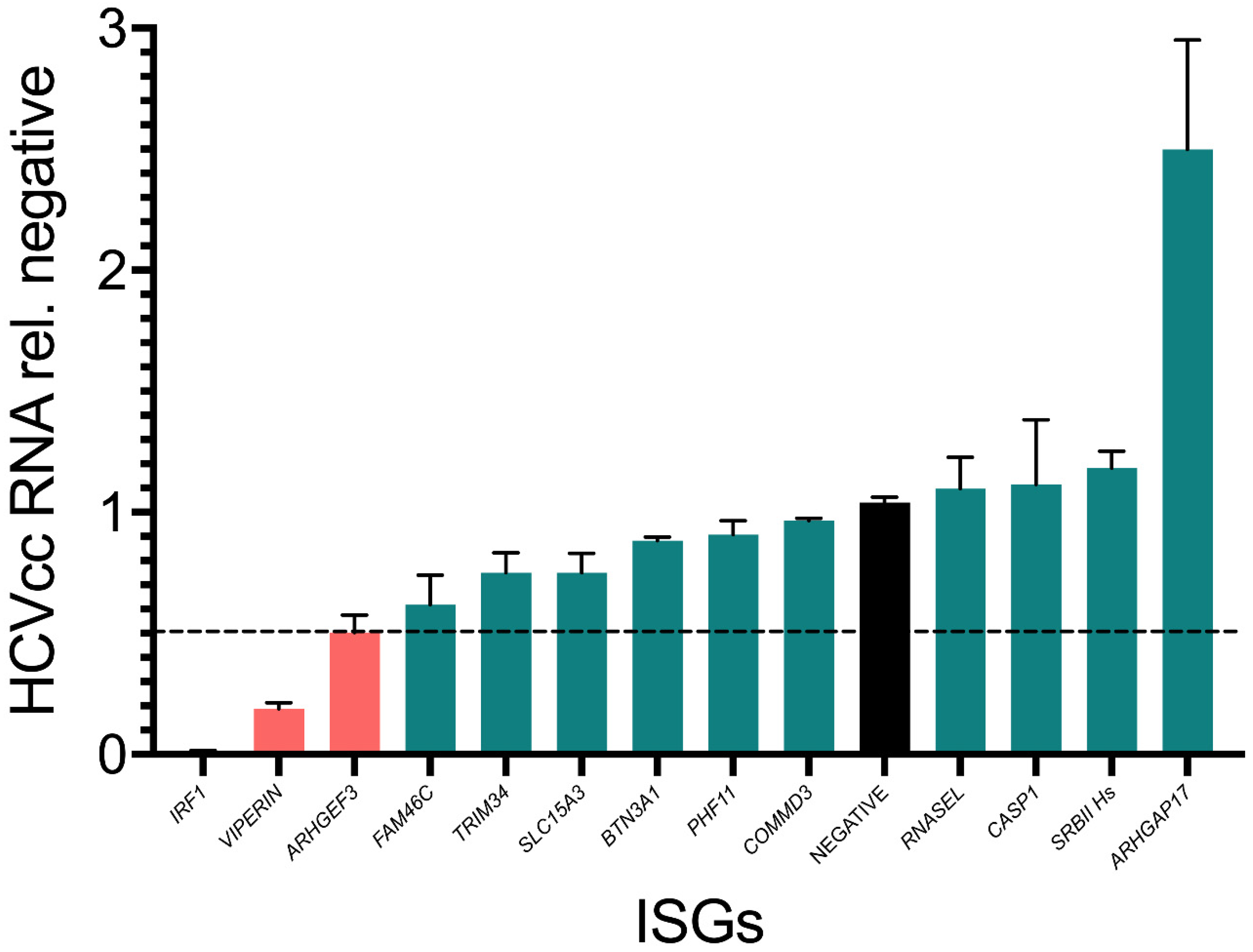
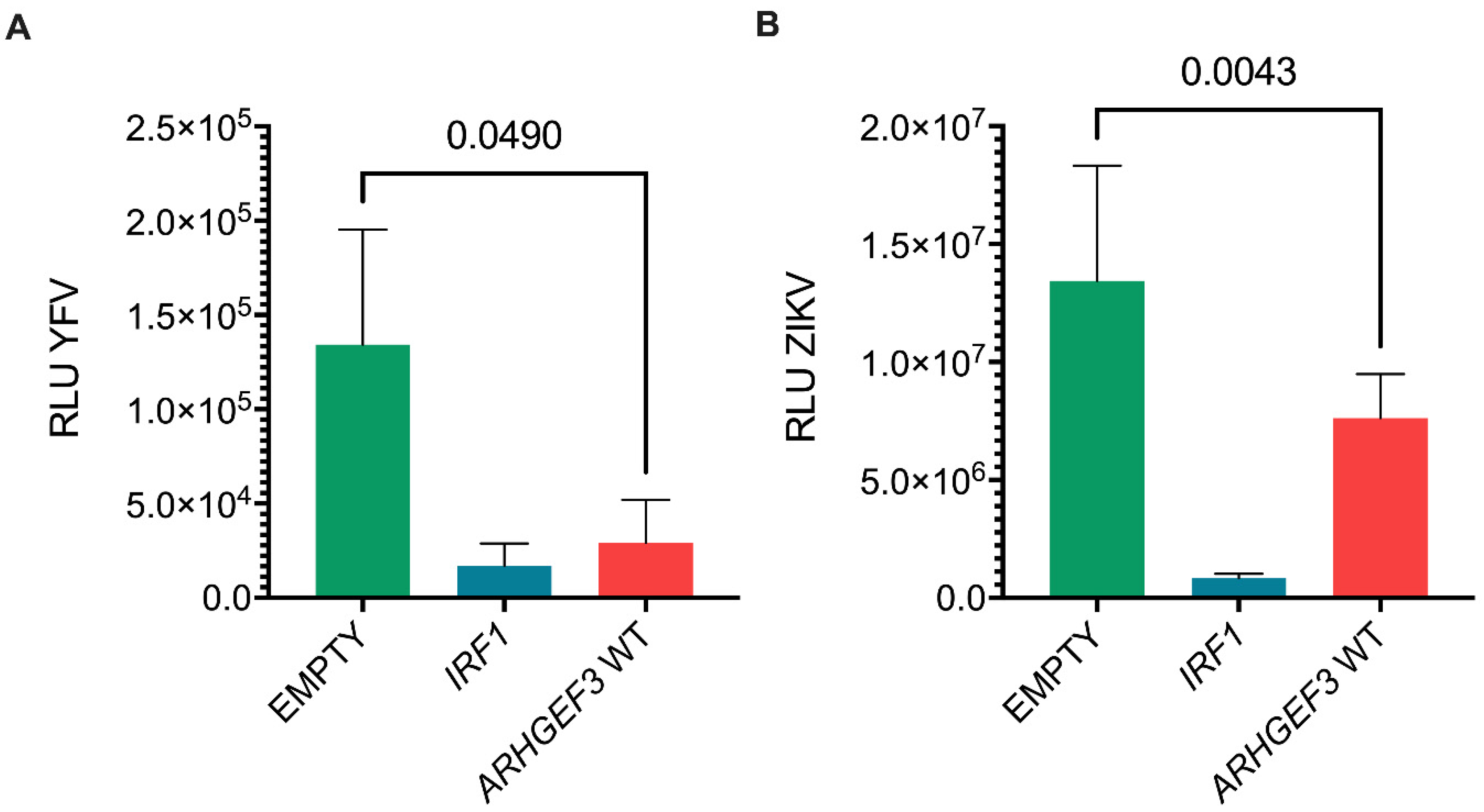
3.3. Assessment of Inhibitor Activity against Replication of Infectious Virus
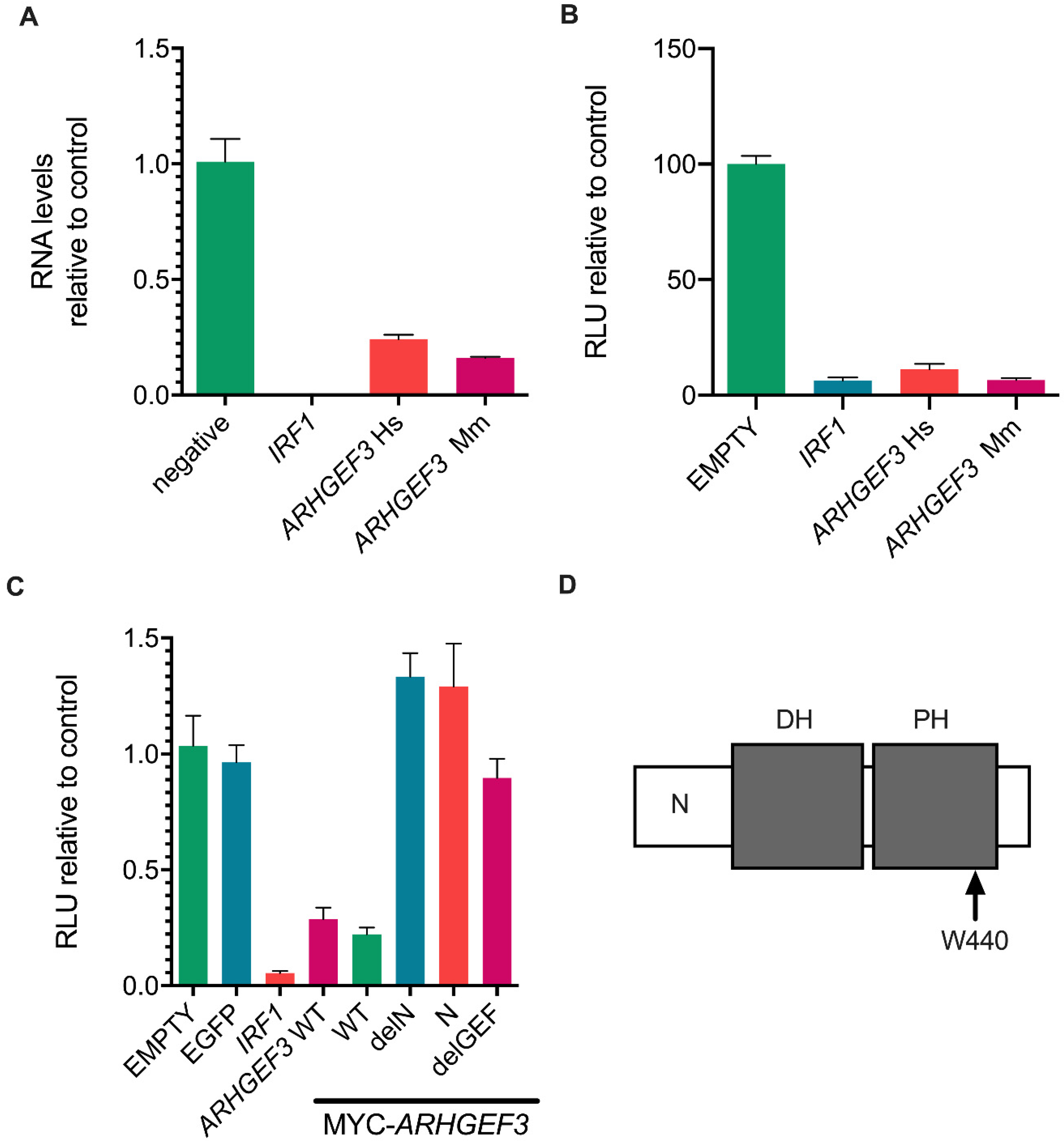
3.4. ARHGEF3 Inhibits HCV Replication
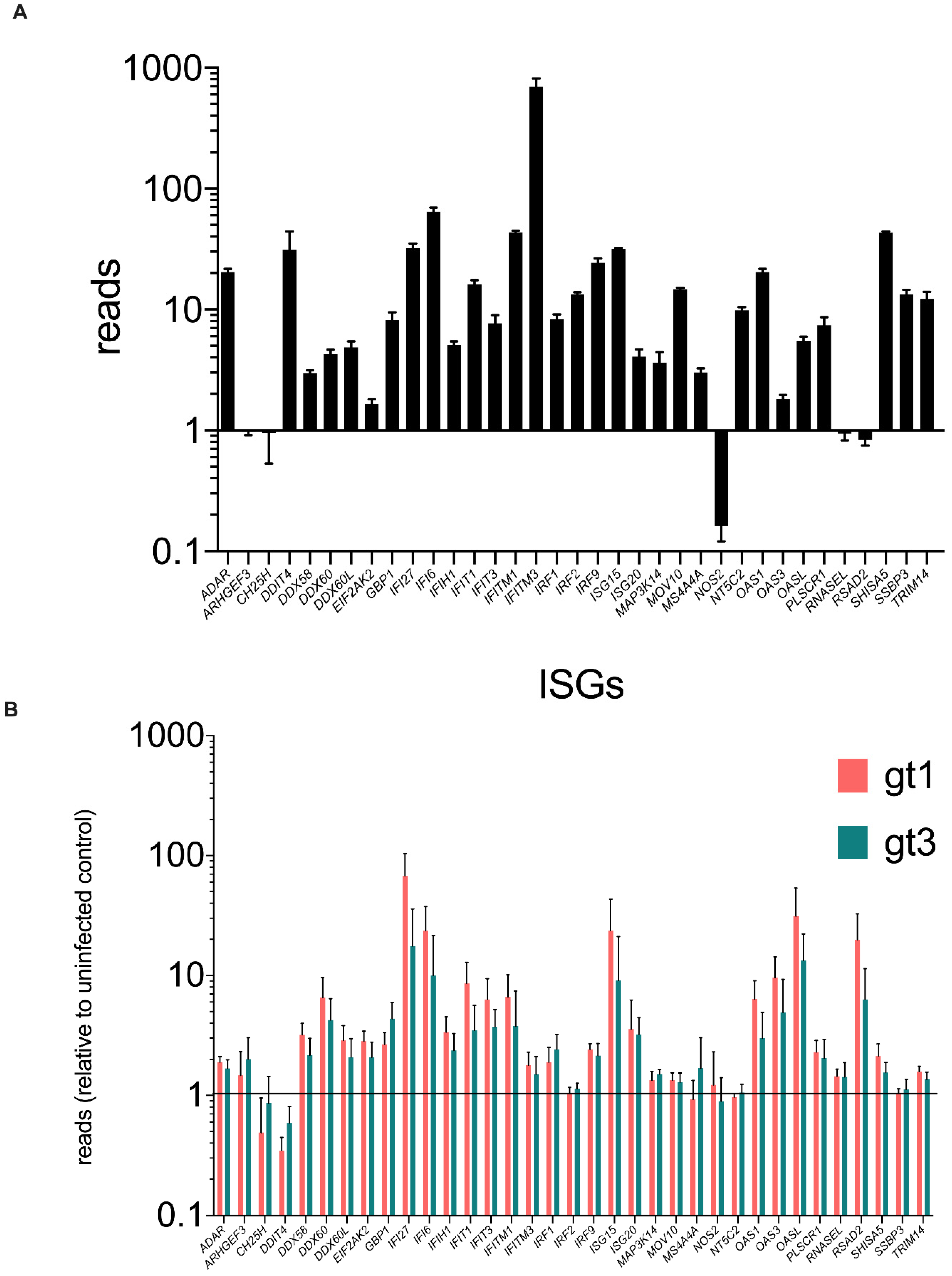
3.5. ARHGEF3 Is Expressed and Modestly Up-Regulated in the Human Liver during Chronic HCV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simmonds, P.; Becher, B.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Hartlage, A.S.; Cullen, J.M.; Kapoor, A. The Strange, Expanding World of Animal Hepaciviruses. Annu. Rev. Virol. 2016, 3, 53–75. [Google Scholar] [CrossRef] [Green Version]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.-M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Prim. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Bigger, C.; Bassett, S.; Klimpel, G. The chimpanzee model of hepatitis C virus infections. ILAR J. 2001, 42, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Bukh, J.; Apgar, C.L.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Failure to infect rhesus monkeys with hepatitis C virus strains of genotypes 1a, 2a or 3a. J. Viral Hepat. 2001, 8, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kurata, T.; Teramoto, Y.; Shiga, J.; Shikata, T. Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J. Med. Primatol. 1993, 22, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, O.; Kochs, G. Mx genes: Host determinants controlling influenza virus infection and trans-species transmission. Hum. Genet. 2020, 139, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017, 15, e2004086. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Serti, E.; Eke, O.; Muchmore, B.; Prokunina-Olsson, L.; Capone, S.; Folgori, A.; Rehermann, B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 2012, 56, 2060–2070. [Google Scholar] [CrossRef]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [Green Version]
- Bamford, C.G.G.; McLauchlan, J. An interferon lambda 4-associated variant in the hepatitis C virus RNA polymerase affects viral replication in infected cells. J. Gen. Virol. 2020, 102, 001495. [Google Scholar] [CrossRef]
- Bamford, C.G.G.; Aranday-Cortes, E.; Cordeiro-Filipe, I.; Sukumar, S.; Mair, D.; da Silva-Filipe, A.; Mendoza, J.L.; Garcia, K.C.; Fan, S.; Tishkoff, S.A.; et al. A polymorphic residue that attenuates the antiviral potential of interferon lambda 4 in hominid lineages. PLoS Pathog. 2018, 14, e1007307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient Initiation of HCV RNA Replication in Cell Culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H. 25 years of interferon-based treatment of chronic hepatitis C: An epoch coming to an end. Nat. Rev. Immunol. 2013, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Gale, M. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013, 19, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.-T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [Green Version]
- Itsui, Y.; Sakamoto, N.; Kurosaki, M.; Kanazawa, N.; Tanabe, Y.; Koyama, T.; Takeda, Y.; Nakagawa, M.; Kakinuma, S.; Sekine, Y.; et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J. Viral Hepat. 2006, 13, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Chu, V.; Wu, N.C.; Chen, Z.; Truong, S.; Brar, G.; Su, S.-Y.; Du, Y.; Arumugaswami, V.; Olson, C.A.; et al. Systematic identification of anti-interferon function on hepatitis C virus genome reveals p7 as an immune evasion protein. Proc. Natl. Acad. Sci. USA 2017, 114, 2018–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Kwon, Y.-C.; Liu, S.; Hagedorn, C.H.; Ray, R.B.; Ray, R. Interferon-α inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci. Rep. 2015, 5, 9012. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Kim, H.; Ray, R.; Ray, R.B. ISG56 and IFITM1 Proteins Inhibit Hepatitis C Virus Replication. J. Virol. 2011, 85, 12881–12889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, C.; Woodward, J.; Lau, D.T.-Y.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Dong, H.; Zhu, H.; Nelsono, D.; Liu, C.; Lambiase, L.; Li, X. Identification of the IFITM3 gene as an inhibitor of hepatitis C viral translation in a stable STAT1 cell line. J. Viral Hepat. 2011, 18, e523–e529. [Google Scholar] [CrossRef] [Green Version]
- Metz, P.; Dazert, E.; Ruggieri, A.; Mazur, J.; Kaderali, L.; Kaul, A.; Zeuge, U.; Windisch, M.P.; Trippler, M.; Lohmann, V.; et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 2012, 56, 2082–2093. [Google Scholar] [CrossRef]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; Narayana, S.; Li, K.; Fiches, G.; McCartney, E.M.; Jangra, R.K.; Lemon, S.M.; Beard, M.R. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef]
- Itsui, Y.; Sakamota, N.; Kakinuma, S.; Nakagawa, M.; Sekine-Osajima, Y.; Tasaka-Fujita, M.; Nishimura-Sakurai, Y.; Suda, G.; Karakama, Y.; Mishima, K.; et al. Antiviral effects of the interferon-induced protein guanylate binding protein 1 and its interaction with the hepatitis C virus NS5B protein. Hepatology 2009, 50, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Anggakusuma; Romera-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.R.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Boys, I.N.; Xu, E.; Mar, K.B.; De La Cruz-Rivera, P.C.; Eitson, J.L.; Moon, B.; Schoggins, J.W. RTP4 Is a Potent IFN-Inducible Anti-flavivirus Effector Engaged in a Host-Virus Arms Race in Bats and Other Mammals. Cell Host Microbe 2020, 28, 712–723.e9. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.; Bamford, C.G.G.; Boutell, C.; McLauchlan, J. Inhibition of hepatitis C virus RNA replication by ISG15 does not require its conjugation to protein substrates by the HERC5 E3 ligase. J. Gen. Virol. 2015, 96, 3236. [Google Scholar] [CrossRef] [PubMed]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Brüggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef]
- Grünvogel, O.; Esser-Nobis, K.; Reustle, A.; Schult, P.; Müller, B.; Metz, P.; Trippler, M.; Windisch, M.P.; Frese, M.; Binder, M.; et al. DDX60L Is an Interferon-Stimulated Gene Product Restricting Hepatitis C Virus Replication in Cell Culture. J. Virol. 2015, 89, 10548–10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.M.; Domingues, P.; Targett-Adams, P.; McLauchlan, J. Comparison of U2OS and Huh-7 cells for identifying host factors that affect hepatitis C virus RNA replication. J. Gen. Virol. 2010, 91, 2238–2248. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.-L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete Replication of Hepatitis C Virus in Cell Culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Royle, J.; Donald, C.L.; Merits, A.; Kohl, A.; Varjak, M. Differential effects of lipid biosynthesis inhibitors on Zika and Semliki Forest viruses. Vet. J. 2017, 230, 62–64. [Google Scholar] [CrossRef]
- Sanchez-Velazquez, R.; de Lorenzo, G.; Tandavanitj, R.; Setthapramote, C.; Bredenbeek, P.J.; Bozzacco, L.; MacDonald, M.R.; Clark, J.J.; Rice, C.M.; Patel, A.H.; et al. Generation of a reporter yellow fever virus for high throughput antiviral assays. Antiviral Res. 2020, 183, 104939. [Google Scholar] [CrossRef]
- Schöfl, G.; Lang, K.; Quenzel, P.; Böhme, I.; Sauter, J.; Hofmann, J.A.; Pingel, J.; Schmidt, A.H.; Lange, V. 2.7 million samples genotyped for HLA by next generation sequencing: Lessons learned. BMC Genom. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiesen, S.; Kübart, S.; Ropers, H.H.; Nothwang, H.G. Isolation of two novel human RhoGEFs, ARHGEF3 and ARHGEF4, in 3p13-21 and 2q22. Biochem. Biophys. Res. Commun. 2000, 273, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Fang, Y.; Yoon, M.-S.; Chen, J. XPLN is an endogenous inhibitor of mTORC2. Proc. Natl. Acad. Sci. USA 2013, 110, 15979–15984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.W.; Aranday-Cortes, E.; Gatherer, D.; Swann, R.; Liefhebber, J.M.; Da Silva Filipe, A.; Sigruener, A.; Barclay, S.T.; Mills, P.R.; Patel, A.H.; et al. Viral genotype correlates with distinct liver gene transcription signatures in chronic hepatitis C virus infection. Liver Int. 2015, 35, 2256–2264. [Google Scholar] [CrossRef] [Green Version]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef]
- Scull, M.A.; Shi, C.; de Jong, Y.P.; Gerold, G.; Ries, M.; von Schaewon, M.; Donovan, B.M.; Labitt, R.N.; Horwitz, J.A.; Gaska, J.M.; et al. Hepatitis C virus infects rhesus macaque hepatocytes and simianized mice. Hepatology 2015, 62, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targett-Adams, P.; McLauchlan, J. Development and characterization of a transient-replication assay for the genotype 2a hepatitis C virus subgenomic replicon. J. Gen. Virol. 2005, 86, 3075–3080. [Google Scholar] [CrossRef]
- Binder, M.; Kochs, G.; Bartenschlager, R.; Lohmann, V. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 2007, 46, 1365–1374. [Google Scholar] [CrossRef]
- Arthur, W.T.; Ellerbroek, S.M.; Der, C.J.; Burridge, K.; Wennerberg, K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J. Biol. Chem. 2002, 277, 42964–42972. [Google Scholar] [CrossRef] [Green Version]
- You, J.S.; Singh, N.; Reyes-Ordonez, A.; Khanna, N.; Bao, Z.; Zhao, H.; Chen, J. ARHGEF3 Regulates Skeletal Muscle Regeneration and Strength through Autophagy. Cell Rep. 2021, 34, 108594. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamford, C.G.G.; Aranday-Cortes, E.; Sanchez-Velazquez, R.; Mullan, C.; Kohl, A.; Patel, A.H.; Wilson, S.J.; McLauchlan, J. A Human and Rhesus Macaque Interferon-Stimulated Gene Screen Shows That Over-Expression of ARHGEF3/XPLN Inhibits Replication of Hepatitis C Virus and Other Flavivirids. Viruses 2022, 14, 1655. https://doi.org/10.3390/v14081655
Bamford CGG, Aranday-Cortes E, Sanchez-Velazquez R, Mullan C, Kohl A, Patel AH, Wilson SJ, McLauchlan J. A Human and Rhesus Macaque Interferon-Stimulated Gene Screen Shows That Over-Expression of ARHGEF3/XPLN Inhibits Replication of Hepatitis C Virus and Other Flavivirids. Viruses. 2022; 14(8):1655. https://doi.org/10.3390/v14081655
Chicago/Turabian StyleBamford, Connor G. G., Elihu Aranday-Cortes, Ricardo Sanchez-Velazquez, Catrina Mullan, Alain Kohl, Arvind H. Patel, Sam J. Wilson, and John McLauchlan. 2022. "A Human and Rhesus Macaque Interferon-Stimulated Gene Screen Shows That Over-Expression of ARHGEF3/XPLN Inhibits Replication of Hepatitis C Virus and Other Flavivirids" Viruses 14, no. 8: 1655. https://doi.org/10.3390/v14081655
APA StyleBamford, C. G. G., Aranday-Cortes, E., Sanchez-Velazquez, R., Mullan, C., Kohl, A., Patel, A. H., Wilson, S. J., & McLauchlan, J. (2022). A Human and Rhesus Macaque Interferon-Stimulated Gene Screen Shows That Over-Expression of ARHGEF3/XPLN Inhibits Replication of Hepatitis C Virus and Other Flavivirids. Viruses, 14(8), 1655. https://doi.org/10.3390/v14081655