
Stabilisation of Viral Membrane Fusion Proteins in Prefusion Conformation by Structure-Based Design for Structure Determination and Vaccine Development


Abstract
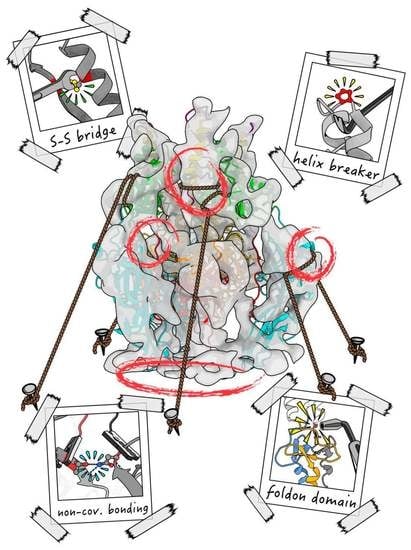
:
1. Introduction
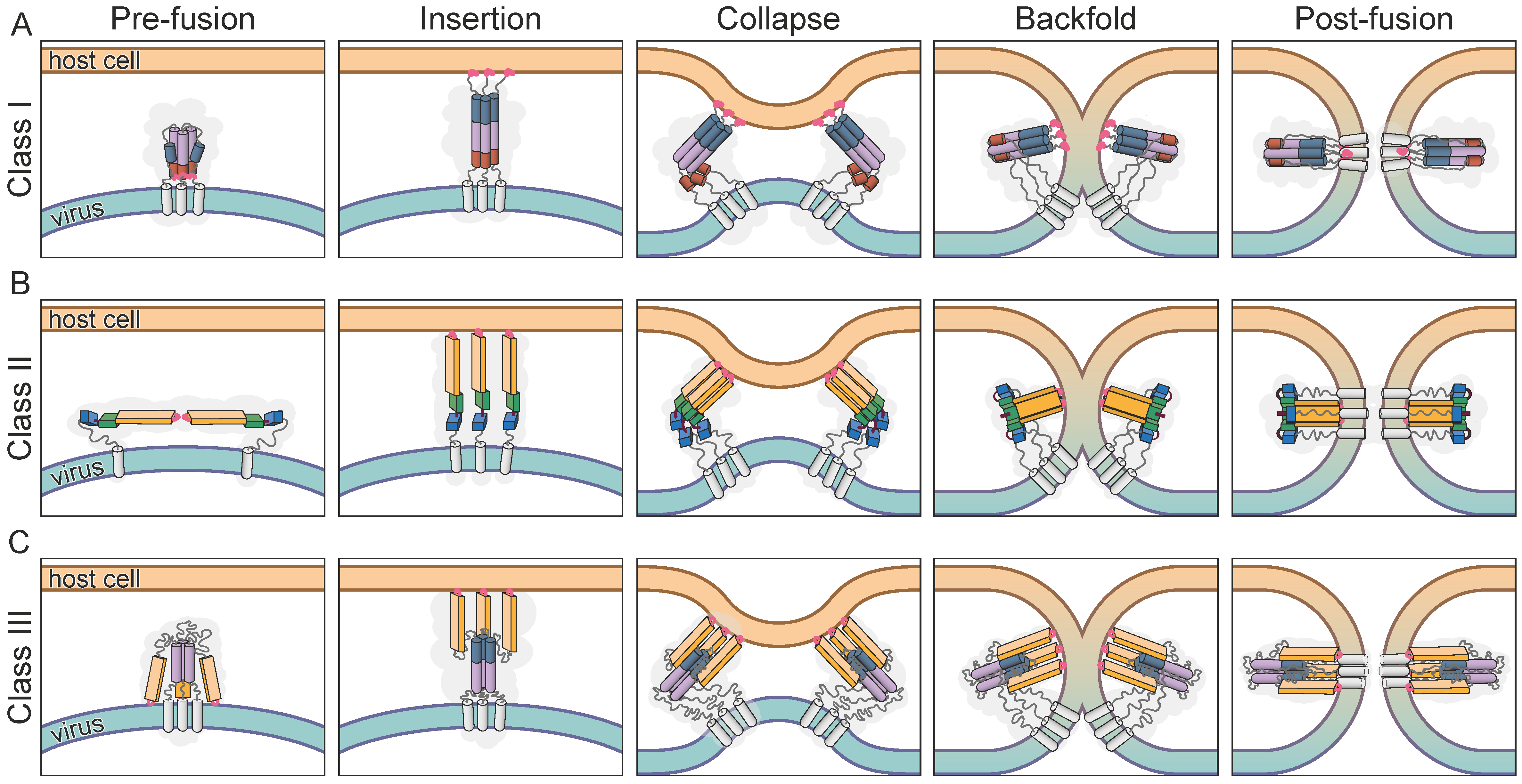
2. The Fusion Process
3. The Classes of Viral Membrane Fusion Proteins
3.1. Class I
3.2. Class II
3.3. Class III
4. Medical Relevance of the Viral Membrane Fusion Proteins
5. The Herpesviral Fusion System
6. Approaches to Stabilise Membrane Fusion Proteins
6.1. Multimerisation Domains

6.2. Disulfide Bonds
6.3. Helix Breaker Insertion
6.4. Cavity Filling
6.5. Charge Balancing
6.6. Combinatorial Mutational Approaches
6.7. Small Molecule Fusion Inhibitors
7. Stabilisation of the gB Prefusion Conformation
7.1. HSV-1 gB
7.2. HCMV gB
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leikin, S.; Parsegian, V.A.; Rau, D.C.; Rand, R.P. Hydration forces. Annu. Rev. Phys. Chem. 1993, 44, 369–395. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479-480, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef]
- Nikolaus, J.; Warner, J.M.; O’Shaughnessy, B.; Herrmann, A. The pathway to membrane fusion through hemifusion. Curr. Top. Membr. 2011, 68, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.V.; Frolov, V.A.; Leikina, E.; Bronk, P.; Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998, 140, 1369–1382. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Schwartz, C.L.; Fischer, E.R.; Ryham, R.J.; Cohen, F.S.; Blank, P.S.; Zimmerberg, J. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 2016, 1, 16050. [Google Scholar] [CrossRef]
- Risselada, H.J.; Grubmuller, H. How proteins open fusion pores: Insights from molecular simulations. Eur. Biophys. J. 2021, 50, 279–293. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef]
- Rey, F.A.; Lok, S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef]
- Benton, D.J.; Gamblin, S.J.; Rosenthal, P.B.; Skehel, J.J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 2020, 583, 150–153. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Roymans, D.; De Bondt, H.L.; Arnoult, E.; Geluykens, P.; Gevers, T.; Van Ginderen, M.; Verheyen, N.; Kim, H.; Willebrords, R.; Bonfanti, J.F.; et al. Binding of a potent small-molecule inhibitor of six-helix bundle formation requires interactions with both heptad-repeats of the RSV fusion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef]
- Mercer, J.; Lee, J.E.; Saphire, E.O.; Freeman, S.A. SnapShot: Enveloped Virus Entry. Cell 2020, 182, 786–786.e781. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. Structural Biology of Influenza Hemagglutinin: An Amaranthine Adventure. Viruses 2020, 12, 1053. [Google Scholar] [CrossRef]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020, 18, e3000626. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The Fusion glycoprotein shell of Semliki Forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- Schibli, D.J.; Weissenhorn, W. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 2004, 21, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Guirakhoo, F.; Heinz, F.X.; Mandl, C.W.; Holzmann, H.; Kunz, C. Fusion activity of flaviviruses: Comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J. Gen. Virol. 1991, 72 Pt 6, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Vaney, M.C.; Roussel, A.; Vigouroux, A.; Reilly, B.; Lepault, J.; Kielian, M.; Rey, F.A. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 2004, 427, 320–325. [Google Scholar] [CrossRef]
- Li, L.; Jose, J.; Xiang, Y.; Kuhn, R.J.; Rossmann, M.G. Structural changes of envelope proteins during alphavirus fusion. Nature 2010, 468, 705–708. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef]
- Steven, A.C.; Spear, P.G. Biochemistry. Viral glycoproteins and an evolutionary conundrum. Science 2006, 313, 177–178. [Google Scholar] [CrossRef]
- Vallbracht, M.; Brun, D.; Tassinari, M.; Vaney, M.C.; Pehau-Arnaudet, G.; Guardado-Calvo, P.; Haouz, A.; Klupp, B.G.; Mettenleiter, T.C.; Rey, F.A.; et al. Structure-Function Dissection of Pseudorabies Virus Glycoprotein B Fusion Loops. J. Virol. 2018, 92, e01203-17. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.L.; Xing, Y.; Chen, D.H.; Roh, S.H.; Pintilie, G.D.; Bushnell, D.A.; Sommer, M.H.; Yang, E.; Carfi, A.; Chiu, W.; et al. A glycoprotein B-neutralizing antibody structure at 2.8 A uncovers a critical domain for herpesvirus fusion initiation. Nat. Commun. 2020, 11, 4141. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, S.; Ciferri, C.; Nikitin, P.A.; Calo, S.; Gerrein, R.; Balabanis, K.; Monroe, J.; Hebner, C.; Lilja, A.E.; Settembre, E.C.; et al. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat. Commun. 2015, 6, 12. [Google Scholar] [CrossRef]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lin, S.; Ye, F.; Yang, J.; Qi, J.; Chen, Z.; Lin, X.; Wang, J.; Yue, D.; Cheng, Y.; et al. Structural Analysis of Rabies Virus Glycoprotein Reveals pH-Dependent Conformational Changes and Interactions with a Neutralizing Antibody. Cell Host Microbe 2020, 27, 441–453.e447. [Google Scholar] [CrossRef] [PubMed]
- Callaway, H.M.; Zyla, D.; Larrous, F.; de Melo, G.D.; Hastie, K.M.; Avalos, R.D.; Agarwal, A.; Corti, D.; Bourhy, H.; Saphire, E.O. Structure of the rabies virus glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Sci. Adv. 2022, 8, eabp9151. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar] [CrossRef]
- Peng, R.; Zhang, S.; Cui, Y.; Shi, Y.; Gao, G.F.; Qi, J. Structures of human-infecting Thogotovirus fusogens support a common ancestor with insect baculovirus. Proc. Natl. Acad. Sci. USA 2017, 114, E8905–E8912. [Google Scholar] [CrossRef]
- Backovic, M.; Jardetzky, T.S. Class III Viral Membrane Fusion Proteins. In Cell Fusion in Health and Disease II: Cell Fusion in Disease; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 714, pp. 91–101. [Google Scholar]
- Liu, Y.; Heim, K.P.; Che, Y.; Chi, X.; Qiu, X.; Han, S.; Dormitzer, P.R.; Yang, X. Prefusion structure of human cytomegalovirus glycoprotein B and structural basis for membrane fusion. Sci. Adv. 2021, 7, eabf3178. [Google Scholar] [CrossRef]
- Si, Z.; Zhang, J.; Shivakoti, S.; Atanasov, I.; Tao, C.L.; Hui, W.H.; Zhou, K.; Yu, X.; Li, W.; Luo, M.; et al. Different functional states of fusion protein gB revealed on human cytomegalovirus by cryo electron tomography with Volta phase plate. PLoS Pathog. 2018, 14, e1007452. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Prazak, V.; Vasishtan, D.; Jefferys, E.E.; Hernandez-Duran, A.; Vallbracht, M.; Klupp, B.G.; Mettenleiter, T.C.; Backovic, M.; Rey, F.A.; et al. The prefusion structure of herpes simplex virus glycoprotein B. Sci. Adv. 2020, 6, eabc1726. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Albertini, A.A.V.; Lepault, J.; Bressanelli, S.; Gaudin, Y. Structures of vesicular stomatitis virus glycoprotein: Membrane fusion revisited. Cell. Mol. Life Sci. 2008, 65, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Oliver, C.; Prince, G.A.; Hemming, V.G.; Pfarr, D.S.; Wang, S.C.; Dormitzer, M.; O’Grady, J.; Koenig, S.; Tamura, J.K.; et al. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar] [CrossRef]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.Y.; et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef]
- Krarup, A.; Truan, D.; Furmanova-Hollenstein, P.; Bogaert, L.; Bouchier, P.; Bisschop, I.J.M.; Widjojoatmodjo, M.N.; Zahn, R.; Schuitemaker, H.; McLellan, J.S.; et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat. Commun. 2015, 6, 8143. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Ranjan, A. The metastable states of proteins. Protein Sci. 2020, 29, 1559–1568. [Google Scholar] [CrossRef]
- Yin, H.S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef]
- Vitu, E.; Sharma, S.; Stampfer, S.D.; Heldwein, E.E. Extensive Mutagenesis of the HSV-1 gB Ectodomain Reveals Remarkable Stability of Its Postfusion Form. J. Mol. Biol. 2013, 425, 2056–2071. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. Distinguishing features of current COVID-19 vaccines: Knowns and unknowns of antigen presentation and modes of action. NPJ Vaccines 2021, 6, 104. [Google Scholar] [CrossRef]
- Israel, A.; Shenhar, Y.; Green, I.; Merzon, E.; Golan-Cohen, A.; Schaffer, A.A.; Ruppin, E.; Vinker, S.; Magen, E. Large-Scale Study of Antibody Titer Decay following BNT162b2 mRNA Vaccine or SARS-CoV-2 Infection. Vaccines 2021, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Moore, J.P. Native-like Env trimers as a platform for HIV-1 vaccine design. Immunol. Rev. 2017, 275, 161–182. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M.; Rabin, L.B.; Feinberg, M.B.; Lieberman, M.; Kosek, J.C.; Reyes, G.R.; Weissman, I.L. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 1988, 53, 55–67. [Google Scholar] [CrossRef]
- Gilbert, P.; Wang, M.; Wrin, T.; Petropoulos, C.; Gurwith, M.; Sinangil, F.; D’Souza, P.; Rodriguez-Chavez, I.R.; DeCamp, A.; Giganti, M.; et al. Magnitude and breadth of a nonprotective neutralizing antibody response in an efficacy trial of a candidate HIV-1 gp120 vaccine. J. Infect. Dis. 2010, 202, 595–605. [Google Scholar] [CrossRef]
- Derking, R.; Sanders, R.W. Structure-guided envelope trimer design in HIV-1 vaccine development: A narrative review. J. Int. AIDS Soc. 2021, 24 (Suppl. S7), e25797. [Google Scholar] [CrossRef]
- Battles, M.B.; Mas, V.; Olmedillas, E.; Cano, O.; Vazquez, M.; Rodriguez, L.; Melero, J.A.; McLellan, J.S. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat. Commun. 2017, 8, 1528. [Google Scholar] [CrossRef]
- Qiao, H.; Pelletier, S.L.; Hoffman, L.; Hacker, J.; Armstrong, R.T.; White, J.M. Specific single or double proline substitutions in the “spring-loaded” coiled-coil region of the influenza hemagglutinin impair or abolish membrane fusion activity. J. Cell Biol. 1998, 141, 1335–1347. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Juraszek, J.; Rutten, L.; Blokland, S.; Bouchier, P.; Voorzaat, R.; Ritschel, T.; Bakkers, M.J.G.; Renault, L.L.R.; Langedijk, J.P.M. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc. Natl. Acad. Sci. USA 2016, 113, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Prussia, A.; Snyder, J.P.; Plemper, R.K. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J. Virol. 2007, 81, 8821–8826. [Google Scholar] [CrossRef] [PubMed]
- Zokarkar, A.; Connolly, S.A.; Jardetzky, T.S.; Lamb, R.A. Reversible inhibition of fusion activity of a paramyxovirus fusion protein by an engineered disulfide bond in the membrane-proximal external region. J. Virol. 2012, 86, 12397–12401. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.G.; Zhang, B.; Ou, L.; Chen, M.; Chuang, G.Y.; Druz, A.; Kong, W.P.; Lai, Y.T.; Rundlet, E.J.; Tsybovsky, Y.; et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat. Struct. Mol. Biol. 2016, 23, 811–820. [Google Scholar] [CrossRef]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar] [CrossRef]
- Kemble, G.W.; Bodian, D.L.; Rose, J.; Wilson, I.A.; White, J.M. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 1992, 66, 4940–4950. [Google Scholar] [CrossRef]
- Slon Campos, J.L.; Marchese, S.; Rana, J.; Mossenta, M.; Poggianella, M.; Bestagno, M.; Burrone, O.R. Temperature-dependent folding allows stable dimerization of secretory and virus-associated E proteins of Dengue and Zika viruses in mammalian cells. Sci. Rep. 2017, 7, 966. [Google Scholar] [CrossRef]
- Rouvinski, A.; Dejnirattisai, W.; Guardado-Calvo, P.; Vaney, M.C.; Sharma, A.; Duquerroy, S.; Supasa, P.; Wongwiwat, W.; Haouz, A.; Barba-Spaeth, G.; et al. Covalently linked dengue virus envelope glycoprotein dimers reduce exposure of the immunodominant fusion loop epitope. Nat. Commun. 2017, 8, 15411. [Google Scholar] [CrossRef]
- Yang, C.; Zeng, F.; Gao, X.; Zhao, S.; Li, X.; Liu, S.; Li, N.; Deng, C.; Zhang, B.; Gong, R. Characterization of two engineered dimeric Zika virus envelope proteins as immunogens for neutralizing antibody selection and vaccine design. J. Biol. Chem. 2019, 294, 10638–10648. [Google Scholar] [CrossRef]
- Slon-Campos, J.L.; Dejnirattisai, W.; Jagger, B.W.; Lopez-Camacho, C.; Wongwiwat, W.; Durnell, L.A.; Winkler, E.S.; Chen, R.E.; Reyes-Sandoval, A.; Rey, F.A.; et al. A protective Zika virus E-dimer-based subunit vaccine engineered to abrogate antibody-dependent enhancement of dengue infection. Nat. Immunol. 2019, 20, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Milder, F.J.; Jongeneelen, M.; Ritschel, T.; Bouchier, P.; Bisschop, I.J.M.; de Man, M.; Veldman, D.; Le, L.; Kaufmann, B.; Bakkers, M.J.G.; et al. Universal stabilization of the influenza hemagglutinin by structure-based redesign of the pH switch regions. Proc. Natl. Acad. Sci. USA 2022, 119, e2115379119. [Google Scholar] [CrossRef] [PubMed]
- Rutten, L.; Gilman, M.S.A.; Blokland, S.; Juraszek, J.; McLellan, J.S.; Langedijk, J.P.M. Structure-Based Design of Prefusion-Stabilized Filovirus Glycoprotein Trimers. Cell Rep. 2020, 30, 4540–4550.e4543. [Google Scholar] [CrossRef]
- Sanders, R.W.; Vesanen, M.; Schuelke, N.; Master, A.; Schiffner, L.; Kalyanaraman, R.; Paluch, M.; Berkhout, B.; Maddon, P.J.; Olson, W.C.; et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 2002, 76, 8875–8889. [Google Scholar] [CrossRef] [PubMed]
- Rutten, L.; Lai, Y.T.; Blokland, S.; Truan, D.; Bisschop, I.J.M.; Strokappe, N.M.; Koornneef, A.; van Manen, D.; Chuang, G.Y.; Farney, S.K.; et al. A Universal Approach to Optimize the Folding and Stability of Prefusion-Closed HIV-1 Envelope Trimers. Cell Rep. 2018, 23, 584–595. [Google Scholar] [CrossRef]
- Hastie, K.M.; Zandonatti, M.A.; Kleinfelter, L.M.; Heinrich, M.L.; Rowland, M.M.; Chandran, K.; Branco, L.M.; Robinson, J.E.; Garry, R.F.; Saphire, E.O. Structural basis for antibody-mediated neutralization of Lassa virus. Science 2017, 356, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; Stewart-Jones, G.B.E.; Tsybovsky, Y.; Caringal, R.T.; Morabito, K.M.; McLellan, J.S.; Chamberlain, A.L.; Nugent, S.T.; Hutchinson, G.B.; Kueltzo, L.A.; et al. Structure-Based Design of Nipah Virus Vaccines: A Generalizable Approach to Paramyxovirus Immunogen Development. Front. Immunol. 2020, 11, 842. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Rush, S.A.; Palomo, C.; Chou, C.W.; Pickens, W.; Mas, V.; McLellan, J.S. Structure-based design of prefusion-stabilized human metapneumovirus fusion proteins. Nat. Commun. 2022, 13, 1299. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Jones, G.B.; Thomas, P.V.; Chen, M.; Druz, A.; Joyce, M.G.; Kong, W.P.; Sastry, M.; Soto, C.; Yang, Y.; Zhang, B.; et al. A Cysteine Zipper Stabilizes a Pre-Fusion F Glycoprotein Vaccine for Respiratory Syncytial Virus. PLoS ONE 2015, 10, e0128779. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef]
- Vallbracht, M.; Backovic, M.; Klupp, B.G.; Rey, F.A.; Mettenleiter, T.C. Common characteristics and unique features: A comparison of the fusion machinery of the alphaherpesviruses Pseudorabies virus and Herpes simplex virus. Adv. Virus Res. 2019, 104, 225–281. [Google Scholar] [CrossRef]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75 Pt 6, 1211–1222. [Google Scholar] [CrossRef]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Banfield, B.W.; Leduc, Y.; Esford, L.; Schubert, K.; Tufaro, F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: Evidence for a proteoglycan-independent virus entry pathway. J. Virol. 1995, 69, 3290–3298. [Google Scholar] [CrossRef]
- Maurer, U.E.; Sodeik, B.; Grunewald, K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. USA 2008, 105, 10559–10564. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V. Herpesvirus Entry into Host Cells Mediated by Endosomal Low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef]
- Dollery, S.J.; Wright, C.C.; Johnson, D.C.; Nicola, A.V. Low-pH-Dependent Changes in the Conformation and Oligomeric State of the Prefusion Form of Herpes Simplex Virus Glycoprotein B Are Separable from Fusion Activity. J. Virol. 2011, 85, 9964–9973. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, S.D.; Lou, H.A.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Structural Basis of Local, pH-Dependent Conformational Changes in Glycoprotein B from Herpes Simplex Virus Type 1. J. Virol. 2010, 84, 12924–12933. [Google Scholar] [CrossRef]
- Komala Sari, T.; Gianopulos, K.A.; Weed, D.J.; Schneider, S.M.; Pritchard, S.M.; Nicola, A.V. Herpes Simplex Virus Glycoprotein C Regulates Low-pH Entry. mSphere 2020, 5, e00826-19. [Google Scholar] [CrossRef]
- Vollmer, B.; Grunewald, K. Herpesvirus membrane fusion—A team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef]
- Guthe, S.; Kapinos, L.; Moglich, A.; Meier, S.; Grzesiek, S.; Kiefhaber, T. Very fast folding and association of a trimerization domain from bacteriophage T4 fibritin. J. Mol. Biol. 2004, 337, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Strelkov, S.V.; Mesyanzhinov, V.V.; Rossmann, M.G. Structure of bacteriophage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain. Structure 1997, 5, 789–798. [Google Scholar] [CrossRef]
- Harbury, P.B.; Kim, P.S.; Alber, T. Crystal structure of an isoleucine-zipper trimer. Nature 1994, 371, 80–83. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Sliepen, K.; van Montfort, T.; Melchers, M.; Isik, G.; Sanders, R.W. Immunosilencing a highly immunogenic protein trimerization domain. J. Biol. Chem. 2015, 290, 7436–7442. [Google Scholar] [CrossRef]
- Mamathambika, B.S.; Bardwell, J.C. Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 2008, 24, 211–235. [Google Scholar] [CrossRef]
- Sanders, R.W.; Moore, J.P. Virus vaccines: Proteins prefer prolines. Cell Host Microbe 2021, 29, 327–333. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, T.; Zhang, Y.; Yang, E.S.; Schramm, C.A.; Shi, W.; Pegu, A.; Oloniniyi, O.K.; Henry, A.R.; Darko, S.; et al. Ultrapotent antibodies against diverse and highly transmissible SARS-CoV-2 variants. Science 2021, 373. [Google Scholar] [CrossRef] [PubMed]
- Vanshylla, K.; Fan, C.; Wunsch, M.; Poopalasingam, N.; Meijers, M.; Kreer, C.; Kleipass, F.; Ruchnewitz, D.; Ercanoglu, M.S.; Gruell, H.; et al. Discovery of ultrapotent broadly neutralizing antibodies from SARS-CoV-2 elite neutralizers. Cell Host Microbe 2022, 30, 69–82.e10. [Google Scholar] [CrossRef]
- Saito, M.; Kono, H.; Morii, H.; Uedaira, H.; Tahirov, T.H.; Ogata, K.; Sarai, A. Cavity-Filling Mutations Enhance Protein Stability by Lowering the Free Energy of Native State. J. Phys. Chem. B 2000, 104, 3705–3711. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yang, P.L. Small-Molecule Inhibition of Viral Fusion Glycoproteins. Annu. Rev. Virol. 2021, 8, 459–489. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, M.J.P.; Kadam, R.U.; Juraszek, J.; Lawson, E.; Brandenburg, B.; Schmitz, F.; Schepens, W.B.G.; Stoops, B.; van Diepen, H.A.; Jongeneelen, M.; et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 2019, 363. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; Langedijk, J.P.; Furmanova-Hollenstein, P.; Chaiwatpongsakorn, S.; Costello, H.M.; Kwanten, L.; Vranckx, L.; Vink, P.; Jaensch, S.; Jonckers, T.H.; et al. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 2016, 12, 87–93. [Google Scholar] [CrossRef]
- Xiao, T.; Frey, G.; Fu, Q.; Lavine, C.L.; Scott, D.A.; Seaman, M.S.; Chou, J.J.; Chen, B. HIV-1 fusion inhibitors targeting the membrane-proximal external region of Env spikes. Nat. Chem. Biol. 2020, 16, 529–537. [Google Scholar] [CrossRef]
- Cooper, R.S.; Georgieva, E.R.; Borbat, P.P.; Freed, J.H.; Heldwein, E.E. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat. Struct Mol. Biol. 2018, 25, 416–424. [Google Scholar] [CrossRef]
- Rogalin, H.B.; Heldwein, E.E. Interplay between the Herpes Simplex Virus 1 gB Cytodomain and the gH Cytotail during Cell-Cell Fusion. J. Virol. 2015, 89, 12262–12272. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Vasishtan, D.; Hernandez Duran, A.; Vollmer, B.; White, P.; Prasad Pandurangan, A.; Siebert, C.A.; Topf, M.; Grunewald, K. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc. Natl. Acad. Sci. USA 2016, 113, 4176–4181. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Vasishtan, D.; Siebert, C.A.; Whittle, C.; Grunewald, K. Extracellular vesicles: A platform for the structure determination of membrane proteins by Cryo-EM. Structure 2014, 22, 1687–1692. [Google Scholar] [CrossRef]
- Jones, T.R.; Lee, S.W.; Johann, S.V.; Razinkov, V.; Visalli, R.J.; Feld, B.; Bloom, J.D.; O’Connell, J. Specific inhibition of human cytomegalovirus glycoprotein B-mediated fusion by a novel thiourea small molecule. J. Virol. 2004, 78, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Spindler, N.; Diestel, U.; Stump, J.D.; Wiegers, A.K.; Winkler, T.H.; Sticht, H.; Mach, M.; Muller, Y.A. Structural Basis for the Recognition of Human Cytomegalovirus Glycoprotein B by a Neutralizing Human Antibody. PLoS Pathog. 2014, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, M.; Lotzsch, H.; Klupp, B.G.; Fuchs, W.; Vollmer, B.; Grunewald, K.; Backovic, M.; Rey, F.A.; Mettenleiter, T.C. In Vitro Viral Evolution Identifies a Critical Residue in the Alphaherpesvirus Fusion Glycoprotein B Ectodomain That Controls gH/gL-Independent Entry. mBio 2021, 12. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Majeed, S.; Ahmad, A.B.; Sehar, U.; Georgieva, E.R. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes 2021, 11, 685. [Google Scholar] [CrossRef] [PubMed]
- Pyle, E.; Zanetti, G. Current data processing strategies for cryo-electron tomography and subtomogram averaging. Biochem. J. 2021, 478, 1827–1845. [Google Scholar] [CrossRef]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Class | Virus | Protein | Mutation(s) | Trimerisation | Cleavage Site Modification | Expression Construct | Fusion Activity | Design Method | PDB Number | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Proline substitution(s) | ||||||||||
| I | hMPV | F | A185P | foldon | ENVRRRR substitution | ectodomain | N.D. | design based on homologous structure (RSV) | 5WB0 | [57] |
| I | Influenza A | HA | V55P, S71P | - | - | full protein | impaired in red blood cell fusion assay | based on structure | - | [58] |
| I | MERS-CoV | S | V1060P, L1061P | foldon | ASVG substitution | ectodomain | impaired in pseudotyped lentivirus infection assays | design based on homologous structures (RSV, HIV, HCoV-HKU1) | 5W9I | [59] |
| I | SARS-CoV | S | K968P, V969P | foldon | - | ectodomain | N.D. | based on structure | 6CRZ | [60] |
| I | SARS-CoV-2 | S | K968P, V969P, A942P, F817P, A892P, A899P | foldon | GSAS substitution | ectodomain | N.D. | based on structure | 6XKL | [61] |
| I | SARS-CoV-2 | S | K986P, V987P | foldon | GSAS substitution | ectodomain | N.D. | based on structure | 6VSB | [62] |
| I | SARS-CoV-2 | S | A892P, A942P, V987P | foldon | GSAS substitution | ectodomain | impaired in cell–cell fusion assay | based on structure | 7A4N, 7AD1 | [63] |
| III | HSV-1 | gB | H516P | - | - | full protein | impaired in cell–cell fusion assay | molecular dynamics simulation | 6Z9M | [43] |
| Cystein-linkage strategy | ||||||||||
| I | HeV | F | Y97C + G131C, N100C + A119C | GCN4 | - | ectodomain | impaired in cell–cell fusion assay | based on structure | - | [64] |
| I | MV | F | I452C + G460C | - | - | full protein | impaired in cell–cell fusion assay | design based on homologous structure (PIV5) | - | [65] |
| I | PIV5 | F | T481C + T482C | - | - | full protein | impaired in cell–cell fusion assay | based on structure | - | [66] |
| I | RSV | F | A149C + Y458C or N183C + N428C | foldon | - | ectodomain | N.D. | based on structure | _ | [67] |
| I | HIV | GP | A501C + T605C | - | LRLRLR substitution | ectodomain | N.D. | mutation screening | - | [68] |
| I | Influenza A | HA | HA1: 212C + 216C | - | - | ectodomain | reduced in red blood cell fusion assay | mutation screening | - | [69] |
| II | DENV | E | A259C or A257C *1 | - | - | ectodomain | N.D. | based on structure | - | [70] |
| II | DENV2 | E | A259C or A257C *1, L107C+A313C | - | - | ectodomain | impaired for double mutant in liposome flotation assay | based on structure | - | [71] |
| II | ZIKV | E | A264C | - | - | ectodomain | impaired in pseudotyped VSV infection assays | design based on homologous structure (DENV) | - | [70,72] |
| II | ZIKV | E | L107C, A264C, A319C, W101A | - | - | ectodomain | N.D., but disrupted fusion loop epitop | design based on homologous structure (DENV) | - | [73] |
| Class | Virus | Protein | Mutation(s) | Trimerisation | Cleavage site modification | Expression construct | Fusion activity | Design method | PDB number | Reference |
| Charge repulsion reduction mutations | ||||||||||
| I | Influenza A | HA | H26W, K51I, E103I | - | - | ectodomain | impaired in cell–cell fusion assay | based on structure | - | [74] |
| Combination of different stabilisation modalities | ||||||||||
| I | Ebola | GP | T577P, K588F | - | *2 | ectodomain | N.D. | design based on homologous structures (RSV, HIV) | 6VKM | [75] |
| I | HIV | GP | I559P, A501C + T605C | - | LRLRLR substitution | ectodomain | N.D. | mutation screening | - | [76] |
| I | HIV | GP | A204I, I573F, K588E, D589V, N651F, K655I, I535N | - | RRRRRR substitution | ectodomain | N.D. | based on structure | 6CK9 | [77] |
| I | LASV | GP | R207C + G360C, E329P | - | RRRR substitution | ectodomain | impaired in pseudotyped VSV infection assays | design based on homologous structures (RSV, HIV) | 5VK2 | [78] |
| I | NiV | F | S191P, L172F, L104C + I114C | GCN4 | - | ectodomain | N.D. | based on structure | - | [79] |
| I | RSV | F | S155C + S290C, S190F, V207L | foldon | - | ectodomain | N.D. | based on structure | 4MMQ–4MMV, 5K6C | [80] |
| I | RSV | F | S215P, N67I | foldon | GSGSGR linker | ectodomain | N.D. | based on structure | 5C6B, 5C69 | [47] |
| I | SARS-CoV | S | A892P, A942P, V987P, D614N, R682S, R685G | foldon | GSA substitution | ectodomain | impaired in cell–cell fusion assay | based on structure | 7A4N, 7AD1 | [63] |
| I | hMPV | F | L110C, T127C, A140C, A147C, N153C, A185P, L219K, V231I, N322C, T365C, N368H, E453Q, V463C | foldon | RRRR substitution | ectodomain | N.D. | design based on structure and homologous structure (RSV) | 7SEM, 7SEJ | [81] |
| Other stabilisation methods | ||||||||||
| I | Influenza A | HA | R329Q (cleavage site mutation) | - | R329Q | full-length | indirect *3 | based on structure and biochem data | - | [82] |
| I | Influenza A | HA | multiple | GCN4 | R329Q | miniHA *4 | N.D. | based on structure | 5CJQ, 5CJS | [83] |
| I | MHV | S | R717S (cleavage site mutation) | GCN4 | R717S | ectodomain | N.D. | biochem data | 3JCL | [84] |
| I | RSV | F | S155C, S290C, S190F, V207L, L512C, L513C, G519C, K520C, M526C, I527C, 533C, 534C, 540C, 541C (cystein Zipper) | foldon | - | ectodomain | N.D. | based on structure | - | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebel, H.; Benecke, T.; Vollmer, B. Stabilisation of Viral Membrane Fusion Proteins in Prefusion Conformation by Structure-Based Design for Structure Determination and Vaccine Development. Viruses 2022, 14, 1816. https://doi.org/10.3390/v14081816
Ebel H, Benecke T, Vollmer B. Stabilisation of Viral Membrane Fusion Proteins in Prefusion Conformation by Structure-Based Design for Structure Determination and Vaccine Development. Viruses. 2022; 14(8):1816. https://doi.org/10.3390/v14081816
Chicago/Turabian StyleEbel, Henriette, Tim Benecke, and Benjamin Vollmer. 2022. "Stabilisation of Viral Membrane Fusion Proteins in Prefusion Conformation by Structure-Based Design for Structure Determination and Vaccine Development" Viruses 14, no. 8: 1816. https://doi.org/10.3390/v14081816
APA StyleEbel, H., Benecke, T., & Vollmer, B. (2022). Stabilisation of Viral Membrane Fusion Proteins in Prefusion Conformation by Structure-Based Design for Structure Determination and Vaccine Development. Viruses, 14(8), 1816. https://doi.org/10.3390/v14081816