
The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
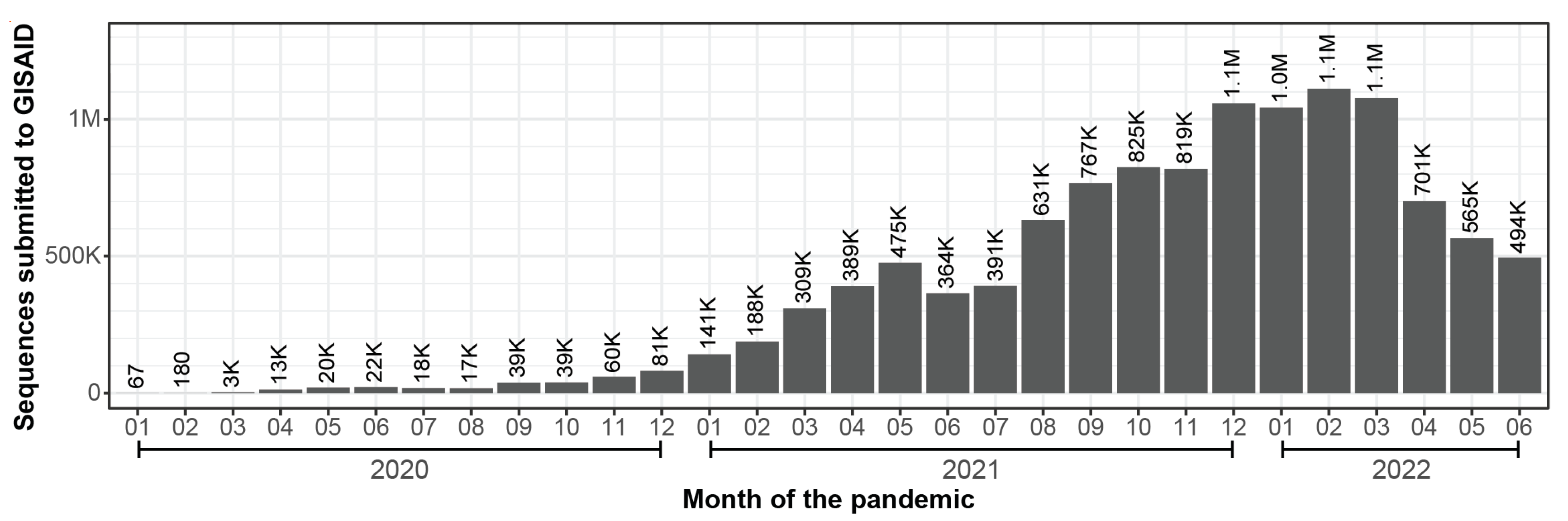
:1. Introduction
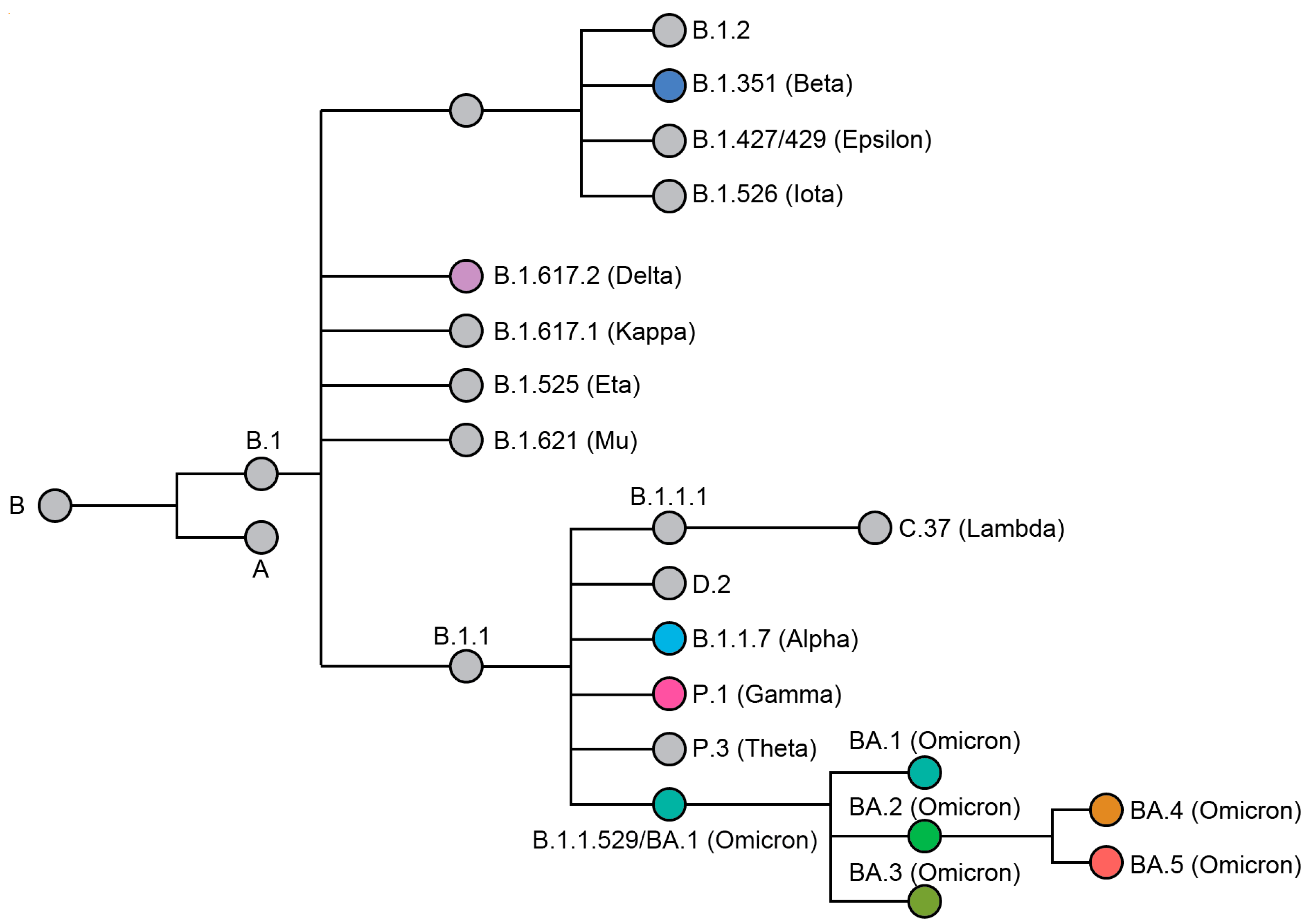
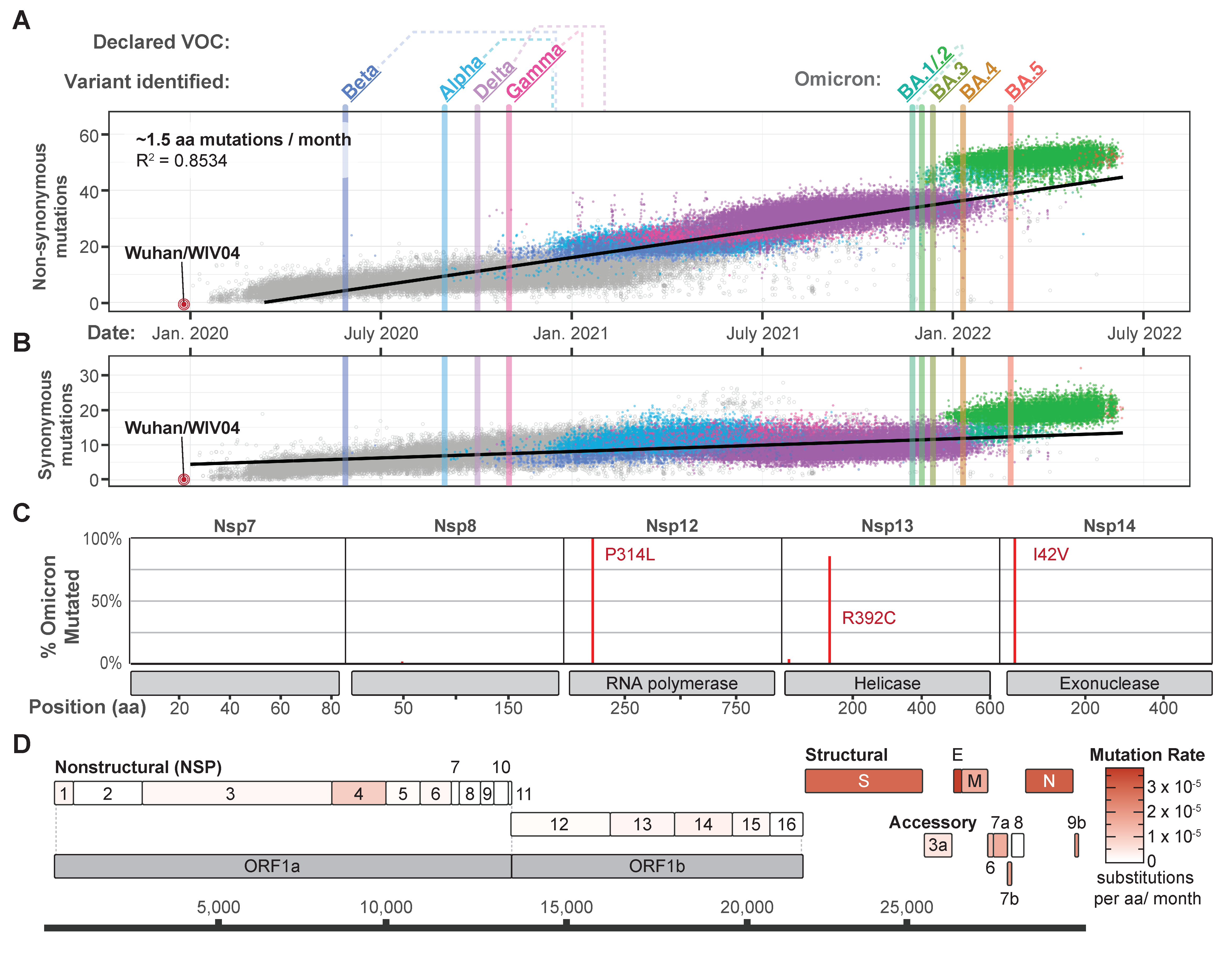
2. Omicron, the Newest Variant of Concern
3. The Future of SARS-CoV-2
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 12 December 2021).
- Callaway, E.; Ledford, H. How bad is Omicron? What scientists know so far. Nature 2021, 600, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Q.; Zhang, W.; Hu, H.; Xu, K. Evidence for selection on SARS-CoV-2 RNA translation revealed by the evolutionary dynamics of mutations in UTRs and CDSs. RNA Biol. 2022, 19, 866–876. [Google Scholar] [CrossRef]
- Ramazzotti, D.; Angaroni, F.; Maspero, D.; Mauri, M.; D’Aliberti, D.; Fontana, D.; Antoniotti, M.; Elli, E.M.; Graudenzi, A.; Piazza, R. Large-scale analysis of SARS-CoV-2 synonymous mutations reveals the adaptation to the human codon usage during the virus evolution. Virus Evol. 2022, 8, veac026. [Google Scholar] [CrossRef]
- Wang, H.; Pipes, L.; Nielsen, R. Synonymous mutations and the molecular evolution of SARS-CoV-2 origins. Virus Evol. 2021, 7, veaa098. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. Available online: https://covariants.org/ (accessed on 18 July 2022).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Quinn, T.P.; Heng, X.; Lorson, C.L.; Sonnerborg, A.; Byrareddy, S.N.; Singh, K. Infectivity of SARS-CoV-2: There Is Something More than D614G? J. Neuroimmune Pharmacol. 2020, 15, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Hsu, J.C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef]
- Moeller, N.H.; Shi, K.; Demir, O.; Belica, C.; Banerjee, S.; Yin, L.; Durfee, C.; Amaro, R.E.; Aihara, H. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc. Natl. Acad. Sci. USA 2022, 119, e2106379119. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020, 11, 5874. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where did ‘weird’ Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Nichols, J.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; Lee, H.; Vanderwood, K.K.; Bimczok, D.; et al. SARS-CoV-2 genomic surveillance identifies naturally occurring truncation of ORF7a that limits immune suppression. Cell Rep. 2021, 35, 109197. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Schneider, T.; Leong, M.; Aravind, L.; Zhang, D. Novel Immunoglobulin Domain Proteins Provide Insights into Evolution and Pathogenesis of SARS-CoV-2-Related Viruses. mBio 2020, 11, e00760-20. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Wang, J.C.; Leelawong, M.; Martin, D.P.; Havens, J.L.; Chowdhury, M.A.; Pekar, J.E.; Amin, H.; Arroyo, A.; Awandare, G.A.; et al. Detection of SARS-CoV-2 intra-host recombination during superinfection with Alpha and Epsilon variants in New York City. Nat. Commun. 2022, 13, 3645. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Fournier, P.E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and identification of a “Deltamicron” SARS-CoV-2 in a three cases cluster in southern France. J. Med. Virol. 2022, 94, 3739–3749. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef]
- Dinnes, J.; Deeks, J.J.; Berhane, S.; Taylor, M.; Adriano, A.; Davenport, C.; Dittrich, S.; Emperador, D.; Takwoingi, Y.; Cunningham, J.; et al. Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection. Cochrane Database Syst. Rev. 2021, 3, CD013705. [Google Scholar] [CrossRef]
- Osterman, A.; Badell, I.; Basara, E.; Stern, M.; Kriesel, F.; Eletreby, M.; Oztan, G.N.; Huber, M.; Autenrieth, H.; Knabe, R.; et al. Impaired detection of omicron by SARS-CoV-2 rapid antigen tests. Med. Microbiol. Immunol. 2022, 211, 105–117. [Google Scholar] [CrossRef]
- Bayart, J.L.; Degosserie, J.; Favresse, J.; Gillot, C.; Didembourg, M.; Djokoto, H.P.; Verbelen, V.; Roussel, G.; Maschietto, C.; Mullier, F.; et al. Analytical Sensitivity of Six SARS-CoV-2 Rapid Antigen Tests for Omicron versus Delta Variant. Viruses 2022, 14, 654. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Hawman, D.W.; Meade-White, K.; Archer, J.; Leventhal, S.S.; Wilson, D.; Shaia, C.; Randall, S.; Khandhar, A.P.; Krieger, K.; Hsiang, T.Y.; et al. SARS-CoV2 variant-specific replicating RNA vaccines protect from disease following challenge with heterologous variants of concern. eLife 2022, 11, e75537. [Google Scholar] [CrossRef] [PubMed]
- Gagne, M.; Moliva, J.I.; Foulds, K.E.; Andrew, S.F.; Flynn, B.J.; Werner, A.P.; Wagner, D.A.; Teng, I.T.; Lin, B.C.; Moore, C.; et al. mRNA-1273 or mRNA-Omicron boost in vaccinated macaques elicits similar B cell expansion, neutralizing responses, and protection from Omicron. Cell 2022, 185, 1556–1571.e1518. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Choi, A.; Koch, M.; Elbashir, S.; Ma, L.; Lee, D.; Woods, A.; Henry, C.; Palandjian, C.; Hill, A.; et al. Variant SARS-CoV-2 mRNA vaccines confer broad neutralization as primary or booster series in mice. Vaccine 2021, 39, 7394–7400. [Google Scholar] [CrossRef]
- Lee, I.J.; Sun, C.P.; Wu, P.Y.; Lan, Y.H.; Wang, I.H.; Liu, W.C.; Yuan, J.P.; Chang, Y.W.; Tseng, S.C.; Tsung, S.I.; et al. A booster dose of Delta × Omicron hybrid mRNA vaccine produced broadly neutralizing antibody against Omicron and other SARS-CoV-2 variants. J. Biomed. Sci. 2022, 29, 49. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Bomfim, C.G.; Barbosa, A.P.; Noda, P.; Noronha, I.L.; Fernandes, B.H.V.; Machado, R.R.G.; Durigon, E.L.; Catanozi, S.; Rodrigues, L.G.; et al. Immunization with SARS-CoV-2 Nucleocapsid protein triggers a pulmonary immune response in rats. PLoS ONE 2022, 17, e0268434. [Google Scholar] [CrossRef] [PubMed]
- Dangi, T.; Class, J.; Palacio, N.; Richner, J.M.; Penaloza MacMaster, P. Combining spike- and nucleocapsid-based vaccines improves distal control of SARS-CoV-2. Cell Rep. 2021, 36, 109664. [Google Scholar] [CrossRef]
- COVID19-Map—John Hopkins Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 21 July 2022).
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention: COVID Data Tracker—Wastewater Surveillance. Available online: https://covid.cdc.gov/covid-data-tracker/#wastewater-surveillance (accessed on 20 July 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiegand, T.; Nemudryi, A.; Nemudraia, A.; McVey, A.; Little, A.; Taylor, D.N.; Walk, S.T.; Wiedenheft, B. The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron. Viruses 2022, 14, 2009. https://doi.org/10.3390/v14092009
Wiegand T, Nemudryi A, Nemudraia A, McVey A, Little A, Taylor DN, Walk ST, Wiedenheft B. The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron. Viruses. 2022; 14(9):2009. https://doi.org/10.3390/v14092009
Chicago/Turabian StyleWiegand, Tanner, Artem Nemudryi, Anna Nemudraia, Aidan McVey, Agusta Little, David N. Taylor, Seth T. Walk, and Blake Wiedenheft. 2022. "The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron" Viruses 14, no. 9: 2009. https://doi.org/10.3390/v14092009
APA StyleWiegand, T., Nemudryi, A., Nemudraia, A., McVey, A., Little, A., Taylor, D. N., Walk, S. T., & Wiedenheft, B. (2022). The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron. Viruses, 14(9), 2009. https://doi.org/10.3390/v14092009