1. Introduction
Enteroviruses (EVs) are a genus of small, non-enveloped, single-stranded positive RNA viruses of the Picornaviridae family and include poliovirus, enterovirus A71 (EV-A71), EV-D68, and coxsackievirus B3 (CVB3) [
1]. CVB3 in particular is the most prevalent etiological pathogen associated with viral myocarditis, which is the most common cause of heart failure and sudden death in infants, adolescents, and young adults [
2,
3,
4]. Despite significant efforts, no clinically validated treatment for this condition is available.
A typical EV lifecycle begins when the viral capsid structure directly binds to a cell entry receptor such as the coxsackievirus and adenovirus receptor (CAR) for CVB3, resulting in the internalization of the virus [
5]. Afterwards, viral RNA is translated into a single large polypeptide, which is subsequently cleaved by two EV-encoded cysteine proteases (2A and 3C) into individual structural and non-structural proteins [
5]. In the meantime, the EV-encoded RNA-dependent RNA polymerase, 3D
pol, replicates the viral genome through a negative strand intermediate to generate positive-stranded viral RNA that is subsequently packaged into icosahedral viral capsids. Eventually, viral progeny is released to infect nearby cells.
EVs are obligate intracellular parasites that rely heavily on the cellular machinery of their hosts to replicate [
1]. The macroautophagy (hereafter referred to as autophagy) pathway is one of these cellular machineries that has been extensively explored. As a result, investigation of the connection between the CVB3 lifecycle and autophagy is warranted. Autophagy is an evolutionarily conserved, fundamental cellular homeostatic pathway by which undesired cytosolic entities such as aggregated proteins, damaged organelles and pathogens are sequestered within double-membraned vesicles (autophagosomes) and sent to lysosomes for degradation [
6]. The biogenesis of the autophagosome begins with the formation of a phagophore, a lipid bilayer that elongates and encloses with the help of a ubiquitin-like conjugation system (ATG5-ATG12-ATG16L) [
7,
8]. This complex recruits the ubiquitin-like, microtubule-associated protein 1A/1B-light chain 3 (LC3) proteins to the developing phagophore and facilitates LC3 attachment to phosphatidylethanolamine (PE) lipids resulting in membrane elongation, curvature, and sealing of the phagophore into a mature autophagosome [
1,
9]. Unlike the sole ATG8 homolog of LC3 in yeast, mammals have two subfamilies: LC3 and γ-aminobutyric acid receptor-associated protein (GABARAP), each encoding three homologs (LC3A, LC3B, LC3C and GABARAP, GABARAPL1, GABARAPL2) [
10]. Among these homologs, LC3B is the most studied [
9]. The processing of LC3 for lipid conjugation is dependent on another ubiquitin-like system, along with the only protease in the ATG protein family, ATG4 [
11,
12]. The ATG4 cysteine protease directly cleaves pro-LC3 (the precursor of LC3) to expose a C-terminal glycine residue to generate a pool of cytosolic LC3-I proteins. Subsequently, LC3-I is recruited to autophagic membranes and directly conjugated in a ubiquitin-like manner with PE lipids, a process termed lipidation [
11,
12]. ATG4 is also responsible for cleaving LC3 from PE, termed delipidation, a process that recycles the cellular pool of LC3 for subsequent autophagosome biogenesis [
12]. In short, ATG4 is an essential component of the autophagy process as it serves a central role in the lipidation and delipidation of LC3, during autophagosome biogenesis. Among the four ATG4 homologs that exist in mammals (ATG4A, ATG4B, ATG4C and ATG4D) [
13], ATG4B was shown to have the most LC3 processing activity as well as the most studied, followed by ATG4A, whereas ATG4C and ATG4D show little activity towards all subtypes of LC3 [
13].
The autophagy pathway is a central component of cell-autonomous innate immunity that selectively targets and degrades various invading viral pathogens following infection. Nevertheless, many studies have reported that EVs can subvert the autophagy process as a viral strategy to evade host innate immunity and facilitate enhanced viral propagation. Most major EVs, such as poliovirus, coxsackievirus, enterovirus A71 and D68, have been shown to stimulate autophagosome formation upon infection, as evidenced by LC3 lipidation and puncta formation, as well as the presence of intracellular double-membraned vesicles [
14,
15]. Intriguingly, recent evidence suggests that EV-induced autophagy is likely a non-canonical process that bypasses the requirements for ULK1/2 and PI3K kinase complexes, components that are essential for the normal autophagy process (e.g., under starvation stress) [
16,
17].
To better understand how EV subverts autophagy, we screened canonical autophagy factors and found out that several proteins were downregulated following EV infection, including ATG4A [
17,
18]. In the current study, we sought to clarify the interaction between EV and ATG4A and determine the role of ATG4A during viral infection using CVB3 as a model enterovirus. We discover that ATG4A is a novel target of viral protease 2A and demonstrate a concentration-dependent effect of ATG4A during CVB3 viral propagation. Furthermore, we show that CVB3 cleaves ATG4A to disrupt its canonical autophagy role.
2. Materials and Methods
2.1. Cell Culture
HeLa and HEK293T cells obtained from American Type Culture Collection (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
2.2. Plasmids
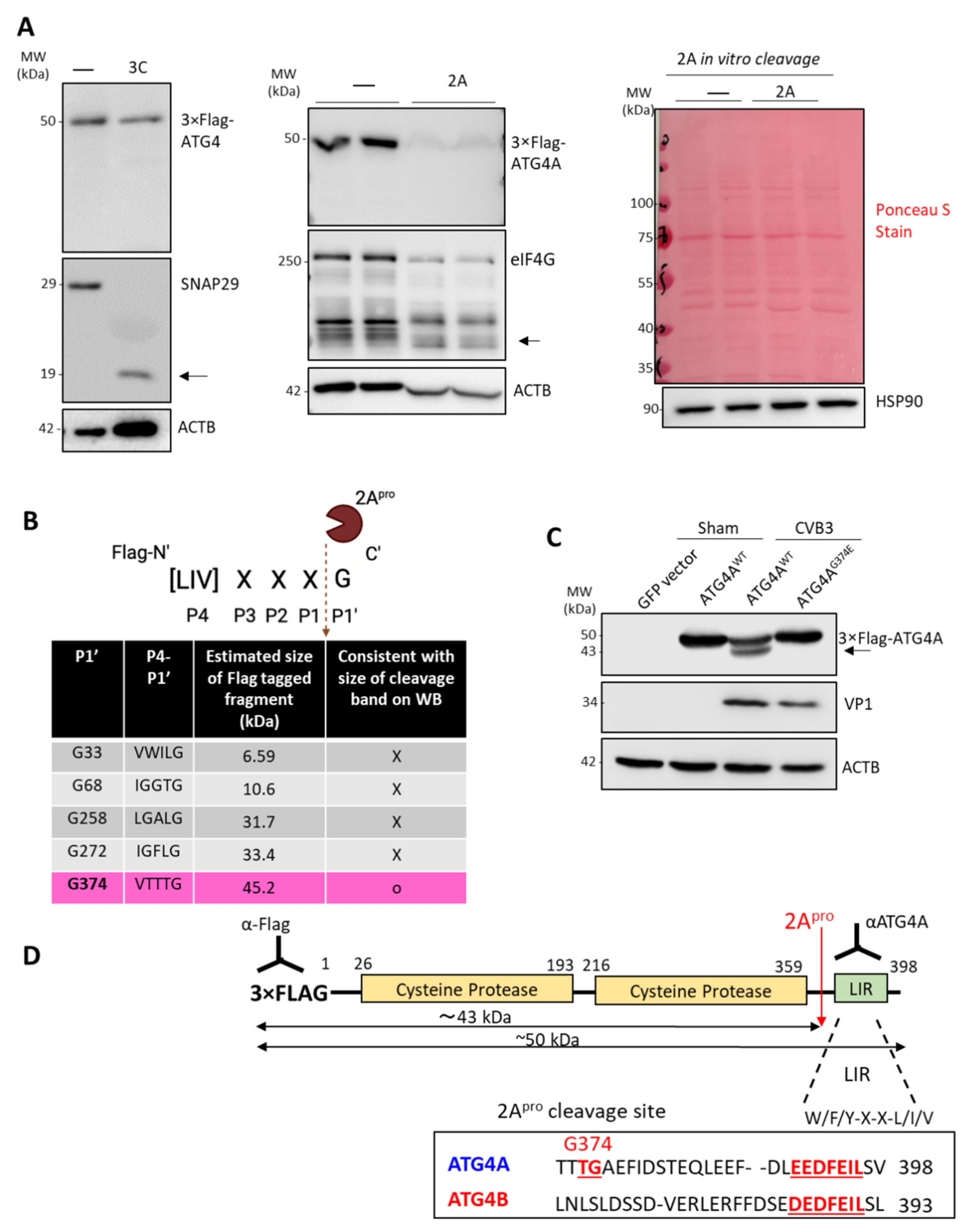
The pEGFP-LC3 plasmid was purchased from addgene (#21073). The 3′Flag-ATG4A (hereby shortened as Flag-ATG4A) and 3′Flag-ATG4B (hereby shortened as Flag-ATG4B) were generated by cloning ATG4A and ATG4B into the multiple cloning sites of p3×Flag CMV10 vector with HindIII/Xbal (ATG4A) and EcoRI/BamHI (ATG4B), respectively. Mutant G374E-ATG4A was generated by site directed mutagenesis and cloning into 3′Flag plasmid. Truncated-ATG4A (deletion at C-terminal after G374) was generated by cloning into 3′Flag plasmid with HindIII and KpnI.
Table 1 summarizes the primer sequences used to amplify each gene.
2.3. Transfections
Plasmid transfections were performed using Lipofectamine 2000 (11668019, Invitrogen) according to the manufacturer’s guidelines. Briefly, 1 mg of DNA diluted in OPTI-MEM (31985062, Thermo Fisher Scientific) was combined with Lipofectamine and incubated at room temperature for 15 min. DNA/lipofectamine mixture was then added dropwise to 6-well plates along with cells (5.5 × 105) for transfection overnight. For RNA interference transfection, Hela cells were transiently transfected with ATG4A siRNA (sc-91197, Santa Cruz Biotechnology) and scrambled siRNA (sc-37007, Santa Cruz Biotechnology) using lipofectamine 2000 following the manufacturer’s instruction. To assess cell viability, siRNA-treated cells were incubated with MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) reagent (Abcam, cat#ab197010) following the manufacturer’s guidelines. Briefly, cells were incubated with 10% MTS solution in complete medium for 2 h at 37 °C. SpectraMax iD3 microplate reader was used to record absorbance at 490 nm of control and treated cells.
2.4. Viral Infections
Seeded cells were infected with CVB3 at different multiplicities of infection (MOI) for various time points as indicated or sham-infected with DMEM. HeLa cells were infected with an MOI = 10 whereas HEK293T cells were infected with an MOI = 100 as previously reported [
17]. Viral titers were quantified as previously described [
19]. Briefly, culture medium from infected samples was serially diluted and transferred to a bed of HeLa cells in 60-well Terasaki plates. After 48 h incubation, 50% tissue culture infective dose titer (TCID50) was calculated by the statistical method of Reed and Muench (HM., 1938 #53). Viral titers are expressed as plaque-forming unit (PFU)/mL with 1 infectious unit equal to 0.7 TCID50.
2.5. Western Blot Analysis
Cells were lysed and harvested with Modified Oncogene Science lysis buffer (MOSLB, 10 mM HEPES with a pH 7.4, 50 mM Na pyrophosphate, 50 mM NaF, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 100 µM Na3VO4, 1% Triton X-100) containing protease inhibitors. Lysates were denatured in 6× sodium dodecyl sulfate protein loading buffer (62.5 mM Tris-HCl at the pH of 6.8, 2% (w/v) SDS, 10% glycerol, 0.01% (w/v) bromophenol blue, and 1.25 M of dithiothreitol). The denatured proteins were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred onto 0.45 μm nitrocellulose membrane for 80 min. at 100 V. For total protein visualization, membranes were stained with Ponceau S (0.1% w/v in 5% acetic acid) and visualized before rinsing with TBST wash buffer. After protein transfer, the membranes were incubated with primary antibodies at 4 °C overnight. The following primary antibodies were used: anti-ATG4A antibody (CST#7613, monoclonal antibody targeting C-terminus, kindly provided by Dr. Sharon Gorski, anti-ATG4B antibody (A2981, Sigma-Aldrich, polyclonal antibody targeting amino acids 6–22), anti-ATG4C antibody (5262, Cell Signaling Technology, polyclonal antibody targeting peptide surrounding Ser430), anti-VP1 antibody (M47, Mediagnost), anti-β-actin antibody (sc-47778, Santa Cruz Biotechnology), anti-LC3B antibody (NB100-2220, Novus Biologicals), anti-Flag antibody (F1804, Sigma-Aldrich), anti-HSP90 (MA110372, Thermofisher). The membranes were then incubated with secondary antibodies for 1 h at room temperature before visualization by chemiluminescence. All primary and secondary antibodies were diluted in 2.5% BSA + Tris-buffered saline with 0.1% Tween® 20 detergent (TBST) with 1:1000 dilutions.
2.6. In Vitro Cleavage Assay
Cell lysates overexpressing Flag-ATG4A were incubated with purified wild-type (WT) CVB3 protease 2A or 3C (0.1 µg) in a cleavage assay buffer (20 mM HEPES pH 7.4, 150 mM potassium acetate, and 1 mM DTT) for 2 h at 37 °C. Reactions were terminated with 6× SDS sample buffer, followed by 95 °C denaturation and subsequent Western blot analysis.
2.7. Real Time Quantitative PCR
Total RNA was extracted using RNeasy Mini Kit (74104, Qiagen). To determine viral gene expression levels, quantitative PCR (qPCR) was conducted with primer pairs targeting viral 2A (forward primer: 5′-GCT TTG CAG ACA TCC GTG ATC-3′; reverse primer: 5′-CAA GCT GTG TTC CAC ATA GTC CTT CA-3′), VP1 (forward primer: 5′-ACA TGG TGC GAA GAG TCT ATT GAG-3′; reverse primer: 5′-TGC TCC GCA GTT AGG ATT AGC-3′), ATG4A (forward primer: 5′-CCA AGC CAG AAG TGA CAA CCAC-3′; reverse primer: 5′-GAC AGA CCT TCA AGT TGA GTT CC-3′) and ACTB (forward primer: 5′-ACT GGA ACG GTG AAG GTG AC-3′; reverse primer: 5′-GTG GAC TTG GGA GAG GAC TG-3′). The qPCR reaction containing 1 μg of RNA was performed using the TaqMan™ RNA-to-CT™ 1-Step Kit (4392653, Thermo Fisher Scientific) on a ViiA 7 Real-Time PCR System (Applied Biosystems). Samples were run in quadruplicate and analyzed using comparative CT (2−ΔΔCT) method with control samples and presented as relative quantitation (RQ). Gene expression of 2A and VP1 was normalized to ACTB mRNA.
2.8. Immunoprecipitation
Immunoprecipitation was performed using anti-FLAG magnetic beads (M8823 Sigma-Aldrich). Briefly, cell lysates were incubated with the beads overnight at 4 °C. Bounded proteins were then eluted with 1% SDS sample buffer and the reactions were terminated by incubating at 95 °C for 10 min. Immunoprecipitated samples were subjected to Western blot analysis.
2.9. Confocal Microscopy
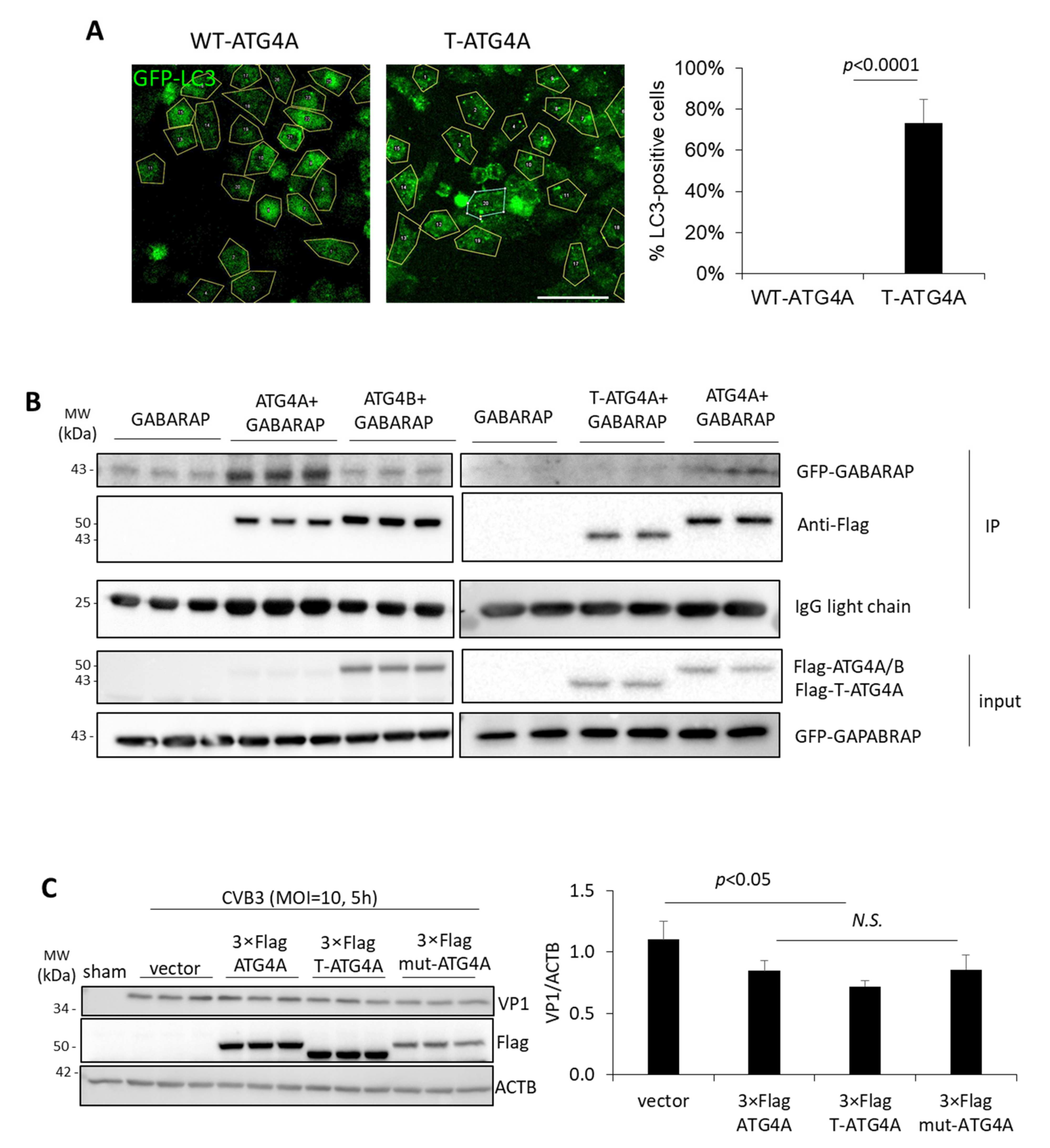
HeLa cells were cultured in 8-well chamber slides (Labtek, 155411) for 24 h. The cells were then transfected with pEGFP-LC3 together with WT-ATG4A or T-ATG4A for an additional 24 h. GFP-LC3 puncta was visualized by Zeiss LSM 880 inverted confocal microscopy and quantified using NIH Image J version 1.53c. Number of GFP-LC3 puncta-positive cells was counted and divided by total cell number to yield the percentage of GFP-LC3 puncta-positive cells (n > 30 cells per condition).
2.10. Statistical Analysis
Densitometric analysis of Western blots was performed by quantifying the intensity of protein bands and normalizing them to ACTB band of sham group unless otherwise noted, using NIH Image J version 1.53c. Each experiment had 3 biological replicates unless otherwise specified. Unpaired student t-test was used for two-group comparison. For multiple group comparisons, one way ANOVA was performed to determine the statistical significance with Tukey’s post-hoc test. Standard deviation was used and p < 0.05 was considered to be statistically significant.
4. Discussion
Despite a miniscule armada of merely eleven viral proteins, EVs are capable of disrupting complex biological systems, such as the cardiovascular and central nervous system, leading to a broad spectrum of human illnesses as minor as the common cold or as severe as viral myocarditis and heart failure [
3,
23]. Among EVs’ arsenals, two virally encoded cysteine proteases, 2A and 3C, are emerging as extremely pathogenic viral factors that disrupt important cellular pathways. The 2A and 3C primarily function to process the viral polyprotein into individual viral proteins or intermediates; however, these proteases routinely target host factors as a strategy to facilitate viral propagation and/or evade host defenses [
24,
25]. Many host substrates of EV proteases have been identified, including several candidates within the autophagy pathway. For example, the autophagy initiating serine-threonine kinase, unc-51-like kinase (ULK1), was recently shown to be targeted by CVB3 to disrupt canonical autophagy signaling. It was demonstrated that viral protease 3C cleaves ULK1 after a central glutamine (Q) 524 residue, resulting in the separation of the N-terminal kinase domain from the C-terminal substrate interaction region and consequently impairing host autophagy regulation in favor of viral pathogenesis [
17]. Therefore, we hypothesized that CVB3 proteases are able to target other autophagy related proteins to further hinder autophagic processes.
Our results demonstrated that CVB3 infection led to the specific downregulation of ATG4A. We discovered that degradation of ATG4A is the consequence of virus-encoded protease 2A-mediated cleavage. The cleavage takes place before G374, which separates the C-terminal kinase domains from the N-terminal LIR. In this study, we focused on the N-terminal cleavage fragment (~43 kDa) as the C-terminal fragment is too small (<3 kDa) to detect by conventional Western blot analysis. Although it is possible that ATG4A may be a collateral target of the promiscuous 2A protease, findings from the current study clearly demonstrate an impaired functional autophagy through abnormalities in LC3 lipidation upon the expression of cleaved T-ATG4A supporting a potential strategy by which virus may disrupt canonical autophagy functions through direct targeting of autophagy factors.
Given that CVB3 targets ATG4A, we then wondered what role ATG4A plays in CVB3 infection. Autophagy can have either pro-viral or anti-viral effect on diverse viruses. For example, Hait et al. [
26] reported that missense mutations in genes encoding ATG4A and LC3B2 disrupt autophagy, resulting in increased herpes simplex virus 2 replication and susceptibility to viral infection in both primary fibroblasts and a neuroblastoma cell line, highlighting the importance of the anti-viral effect of autophagy. Conversely, EVs, such as poliovirus, were shown to usurp autophagy for pro-viral functions, likely by recruiting host (autophagosome) membrane for replication [
27]. In our study, endogenous ATG4A was shown to facilitate viral propagation. We observed both reduced viral RNA and protein expression following ATG4A knockdown, suggesting the necessity of ATG4A presence in viral growth, yet its precise mode of action remains unknown. We wondered why CVB3 would cleave a pro-viral protein. One possibility is that the timing of cleavage could play a role given that ATG4A processing began 5 h post infection, a time point that is considered to be late stage of the viral lifecycle in HeLa cells [
28]. Given that viral-induced double membrane replication organelles appear early, it is plausible that non-cleaved ATG4A exerts its pro-viral effect at an early stage as it facilitates autophagosome biogenesis that can serve as replication compartments for CVB3 [
28,
29]. Additionally, the pro-viral role of ATG4A may be autophagy independent. One possibility is that CVB3 may co-opt the cysteine protease function of ATG4A, similar to the virally encoded cysteine proteases 2A and 3C, to ultimately support viral maturation.
Previous studies have shown that pro-viral factors can be targeted by viral proteases during late infection [
17,
19]. Interestingly, overexpression of exogenous ATG4A is associated with significant reduction in viral protein production although no changes in viral RNA synthesis were observed, signifying that this overexpressed anti-viral function of ATG4A is not viral RNA dependent. Instead, higher concentrations of ATG4A could be acting upon viral proteins or has an effect on other cellular pathways that affect viral protein synthesis (off-target effect). Interestingly, the transfection of a non-cleavable mutant (G374E) or truncated ATG4A, did not significantly impact viral protein production as compared to wildtype ATG4A expression although all three constructs showed a downtrend in viral protein compared to vector only transfected cells, suggesting a general anti-viral role of ATG4A mutants used in the current study. Of note, overexpression of ATG4A significantly blocks CVB3-induced lipidation likely through its delipidation function. We posit that the anti-viral effects of exogenous ATG4A may be the result of its delipidation function and a primary reason why viral protease 2A may disrupt this function through ATG4A cleavage. Additionally, a large amount of exogenous ATG4A may suppress viral replication by potentially accelerating the recycling, or delipidation, of LC3-II. Taken together, these divergent observations suggest that ATG4A may have pro-viral functions during the early stages but anti-viral effects during late infection, providing a plausible explanation of viral-mediated cleavage of ATG4A during late infection.
In this study, we showed that the virally mediated cleavage of the LIR domain of ATG4A leads to impaired autophagy function, including reduced LC3 delipidation and GABARAP interaction. Interestingly, the overexpression of WT-ATG4A is associated with a significant reduction in LC3 puncta whereas approximately 70% of cells expressing T-ATG4A exhibit accumulation of LC3 puncta. Increased LC3 puncta can be interpreted as either an enhancement in the autophagosome biogenesis process or a disruption of the autophagic recycling machinery, resulting in reduced clearance of LC3. We rationalize that ATG4A overexpression is likely enhancing the delipidation process whereas overexpression of T-ATG4A results in the accumulation of LC3 puncta due to an impaired LC3-II delipidation process. The truncation of ATG4A may be a viral strategy to retain autophagosome membranes and indirectly prevent lysosomal fusion. Indeed, EVs have previously been shown to utilize viral protease to disrupt the autophagosome-lysosome fusion process [
21,
30]. In the current study, we utilized a Flag-tagged ATG4A, and we cannot exclude the possibility that 3×Flag tag has other impacts. However, in our previous research, the use of a 3×Flag tag did not interfere with protein function likely due to the small molecular size (~3 kDa) of the tag [
21].
In addition to impaired LC3 interaction, we also showed decreased physical association between T-ATG4A and GABARAP. ATG4A has been reported to have high affinity and catalytic efficiency toward GABARAP, which was confirmed in our interaction assay [
13]. Notably, GABARAP is preferentially involved in membrane tethering and fusion [
10]. It is therefore postulated that cleavage of ATG4A may hinder the ability of GABARAP to promote autophagosome-lysosome fusion, and consequent viral protein degradation.
In summary, our study identified ATG4A as a novel pro-viral host factor that is co-opted by EV protease to impair host autophagy. To our knowledge, this is the first reported cleavage of a host protease by a Picornaviridae viral protease. The cleavage poses one of the underlying mechanisms by which EVs evade autophagy.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}