
Role of Caspases and Gasdermin A during HSV-1 Infection in Mice
 and
and 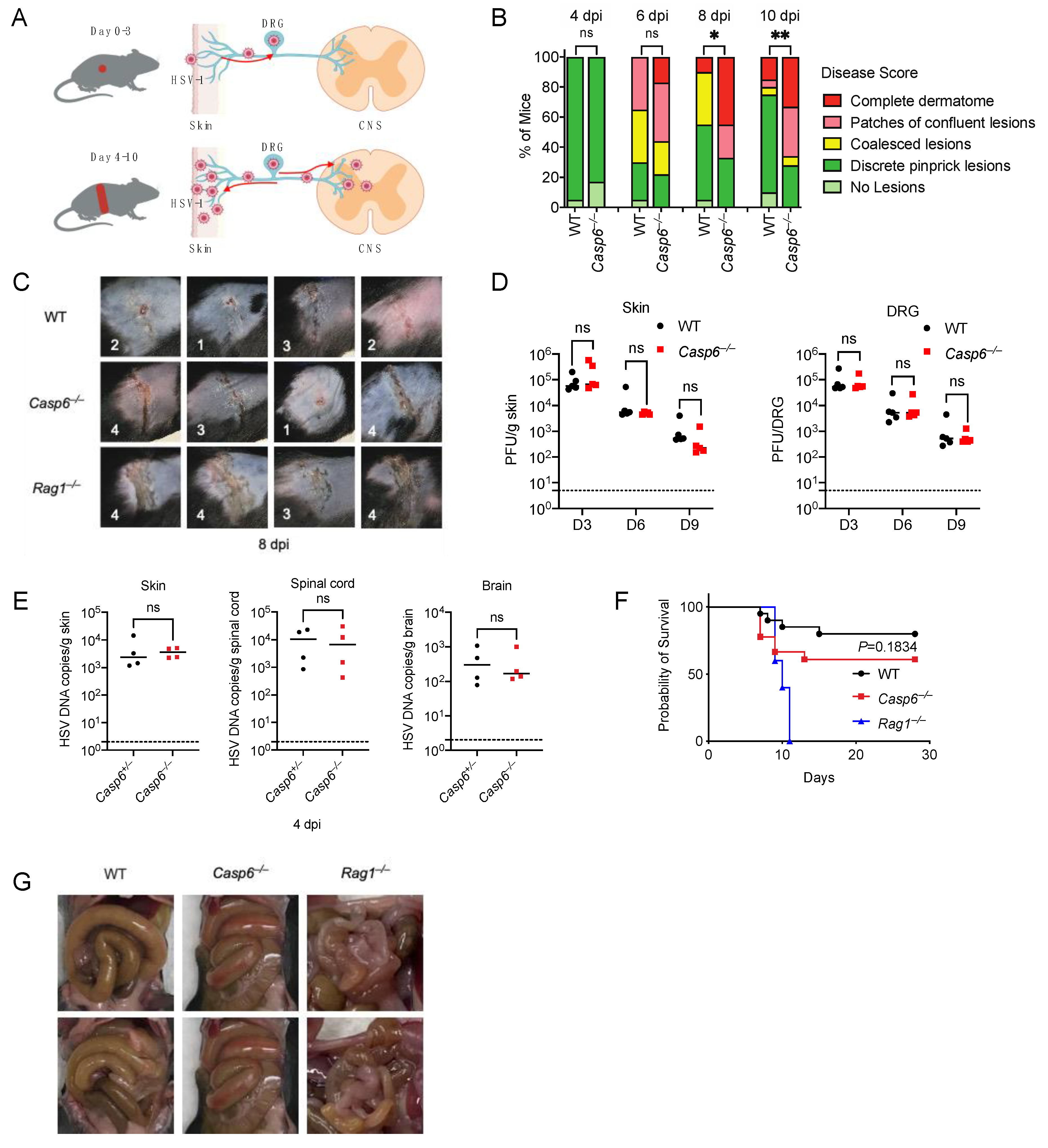{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells and Viruses
2.3. Mouse Flank Scarification Model
2.4. Mouse Footpad Model
2.5. Mouse Corneal Model
2.6. Viral Titers
2.7. Western Blot
2.8. Histological Staining
2.9. Statistics
3. Results
3.1. Caspase-6 Is Partially Required to Defend against HSV-1 Skin Infection
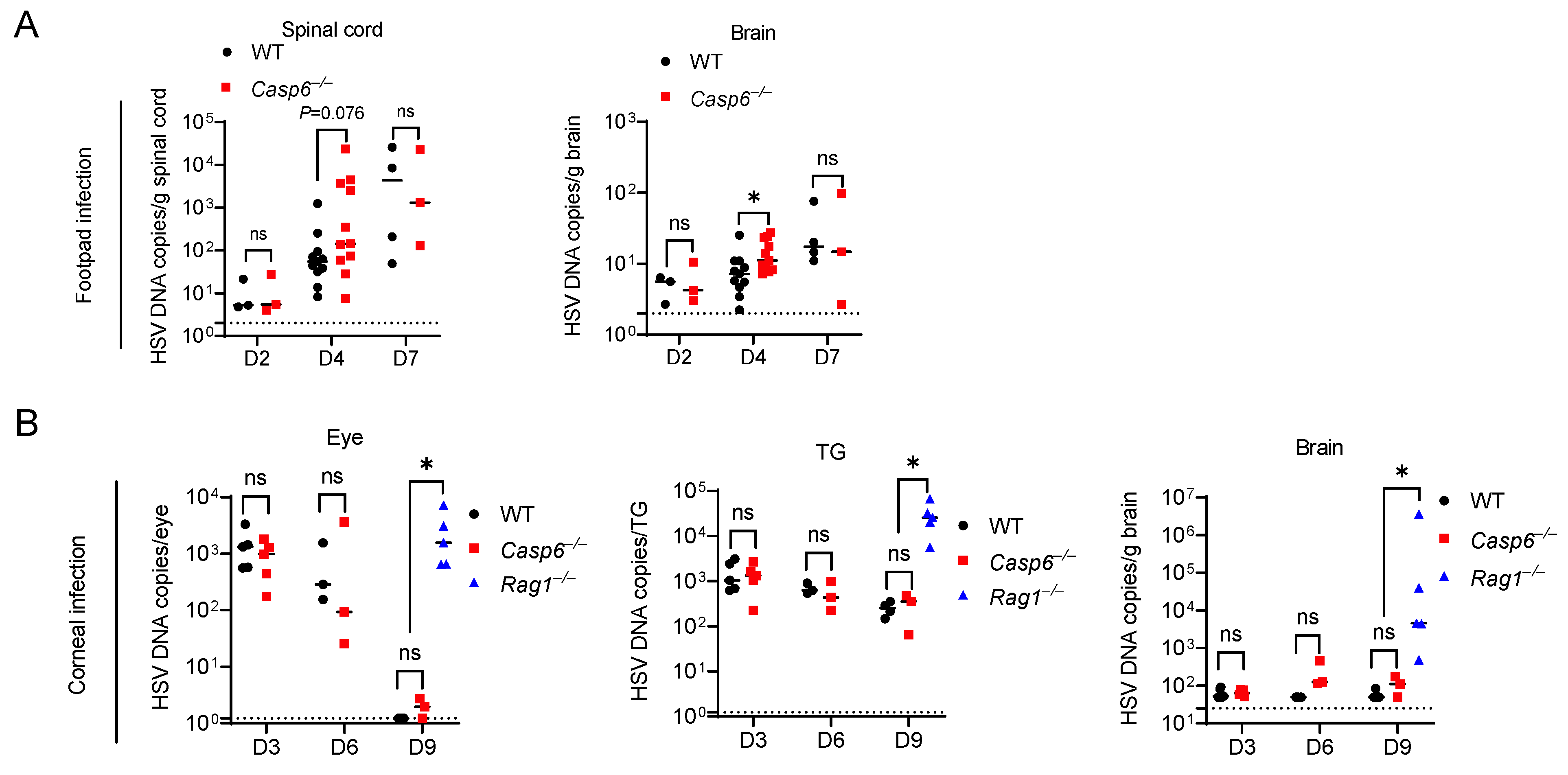
3.2. Caspase-6 Is Partially Required to Control HSV-1 Footpad Infection
3.3. Caspase-6 Is Dispensable for Control of HSV-1 Corneal Infection
3.4. Caspase-6 Is Not Required to Defend against HSV-1 ΔUs3 Strain Infection
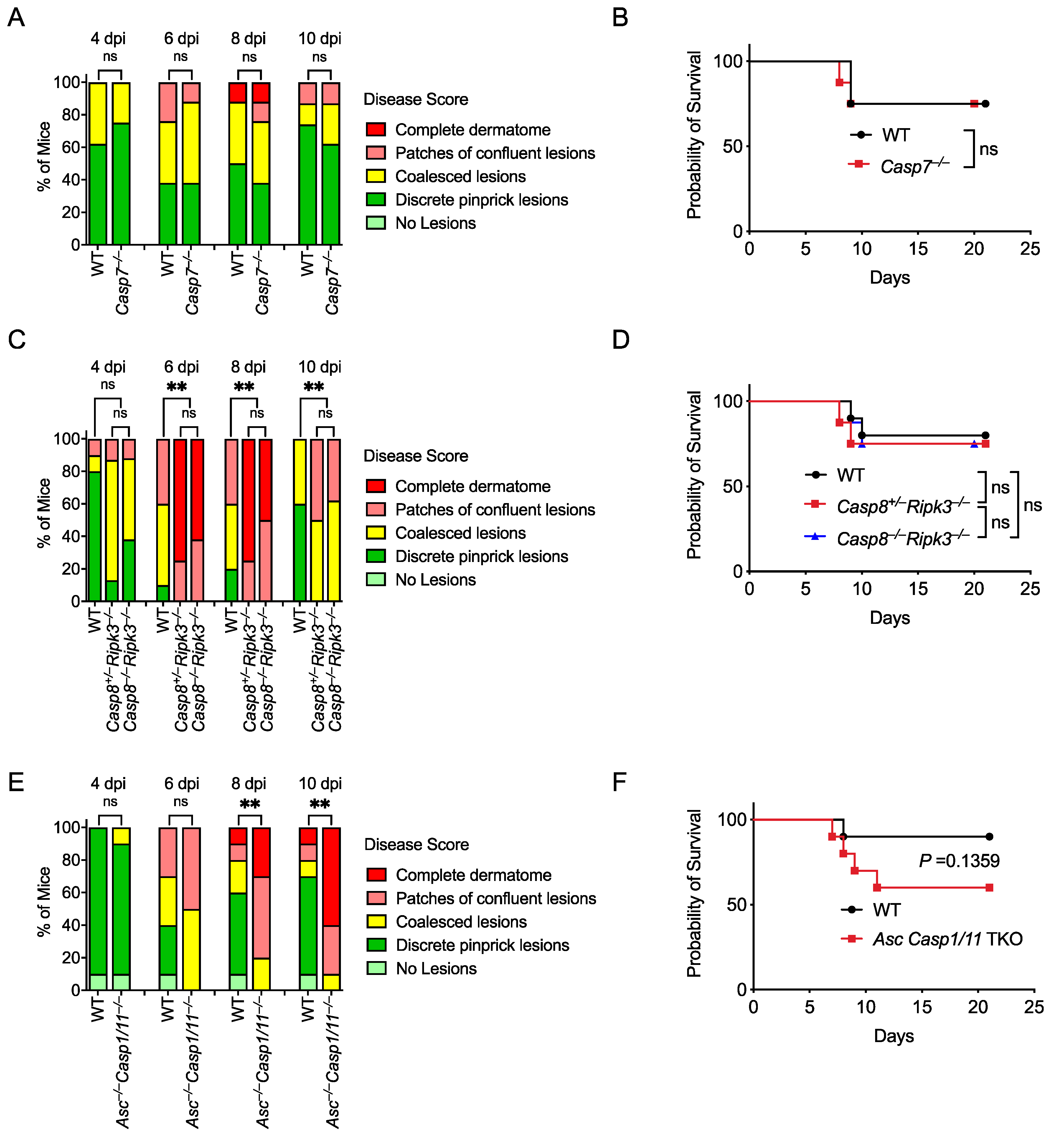
3.5. Caspase-7 Is Not Required to Defend against HSV-1 Skin Infection
3.6. Caspase-8 Is Not Required for Defense against HSV-1, but Ripk3 Is Important
3.7. Asc–/–Casp1/11–/– Mice Develop More Severe Skin Lesions
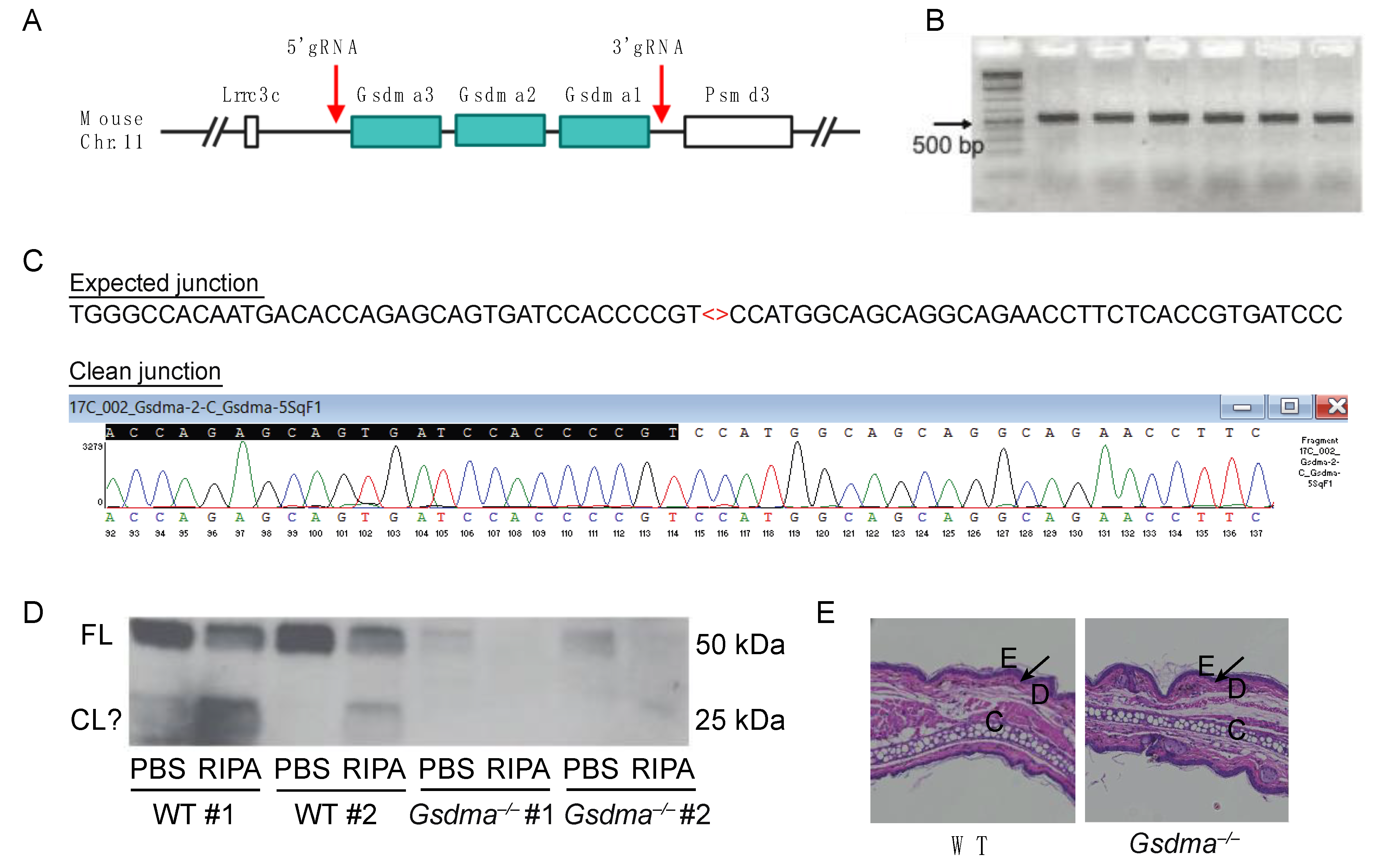
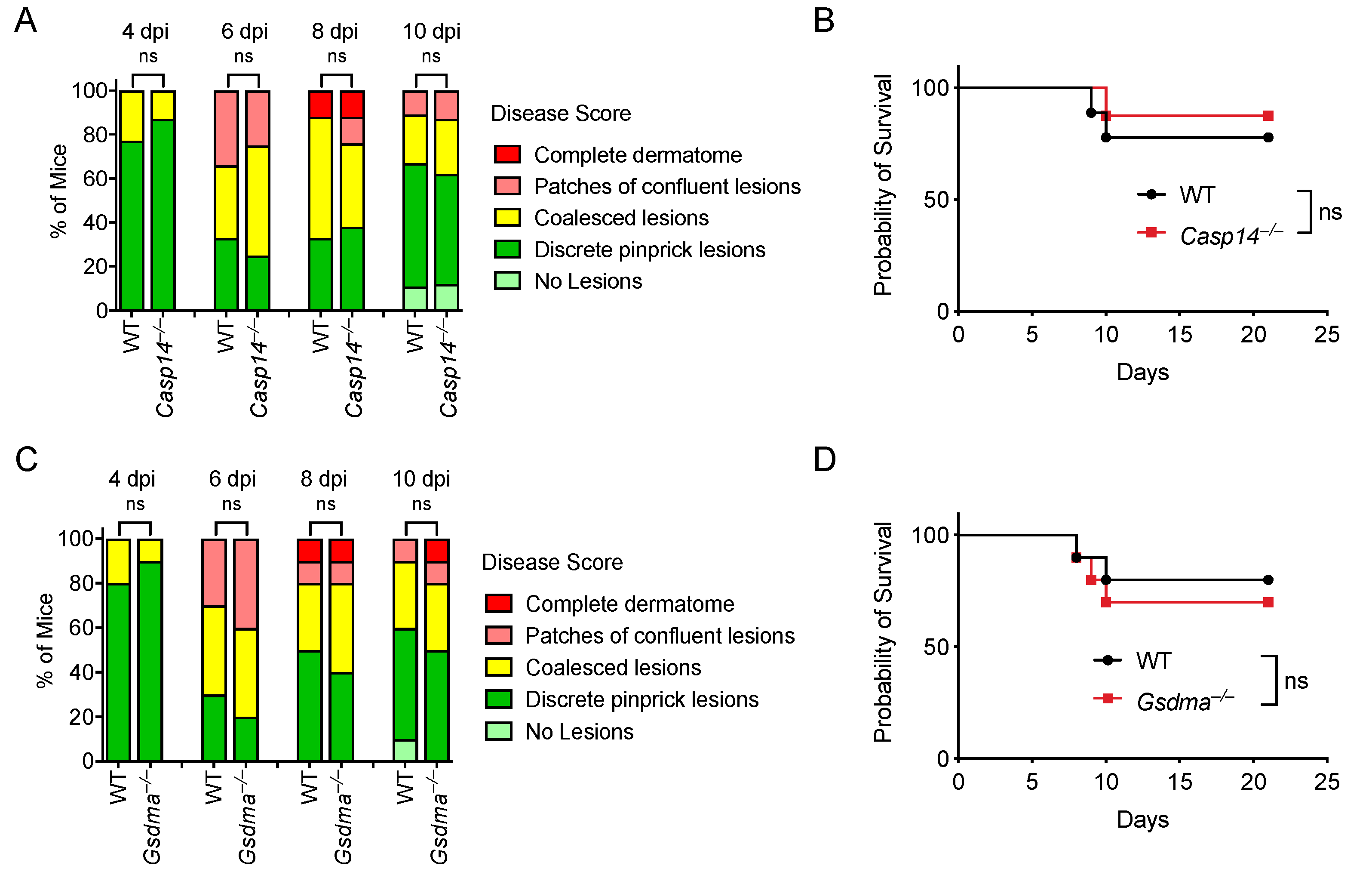
3.8. Caspase-14 and Gasdermin A Are Not Required for Defense against HSV-1 Skin Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Herpes Simplex Virus. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 2 August 2022).
- Xu, F.; Markowitz, L.E.; Gottlieb, S.L.; Berman, S.M. Seroprevalence of herpes simplex virus types 1 and 2 in pregnant women in the United States. Am. J. Obstet. Gynecol. 2007, 196, e1–e43. [Google Scholar] [CrossRef]
- Denes, C.E.; Everett, R.D.; Diefenbach, R.J. Herpes Simplex Virus, Methods and Protocols. Methods Mol. Biology 2019, 2060, 1–30. [Google Scholar]
- van Lint, A.; Ayers, M.; Brooks, A.G.; Coles, R.M.; Heath, W.R.; Carbone, F.R. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J. Immunol. 2003, 172, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A.; Nash, A.A. Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J. Virol. 1984, 52, 816–821. [Google Scholar] [CrossRef]
- McGraw, H.M.; Awasthi, S.; Wojcechowskyj, J.A.; Friedman, H.M. Anterograde Spread of Herpes Simplex Virus Type 1 Requires Glycoprotein E and Glycoprotein I but Not Us9. J. Virol. 2009, 83, 8315–8326. [Google Scholar] [CrossRef]
- Lubinski, J.M.; Lazear, H.M.; Awasthi, S.; Wang, F.; Friedman, H.M. The Herpes Simplex Virus 1 IgG Fc Receptor Blocks Antibody-Mediated Complement Activation and Antibody-Dependent Cellular Cytotoxicity In Vivo. J. Virol. 2011, 85, 3239–3249. [Google Scholar] [CrossRef]
- Engel, J.P.; Madigan, T.C.; Peterson, G.M. The transneuronal spread phenotype of herpes simplex virus type 1 infection of the mouse hind footpad. J. Virol. 1997, 71, 2425–2435. [Google Scholar] [CrossRef]
- Fensterl, V.; Wetzel, J.L.; Sen, G.C.; Lyles, D.S. Interferon-Induced Protein Ifit2 Protects Mice from Infection of the Peripheral Nervous System by Vesicular Stomatitis Virus. J. Virol. 2014, 88, 10303–10311. [Google Scholar] [CrossRef]
- Koganti, R.; Yadavalli, T.; Shukla, D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 2019, 7, 429. [Google Scholar] [CrossRef]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses 2020, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stavrakis, G.; Karaba, A.H. Herpesviruses and Inflammasomes: One Sensor Does Not Fit All. mBio 2022, 13, e01737-21. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Li, L.; Miao, E.A. Innate Sensors Trigger Regulated Cell Death to Combat Intracellular Infection. Annu. Rev. Immunol. 2022, 40, 469–498. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Nozaki, K.; Maltez, V.I.; Rayamajhi, M.; Tubbs, A.L.; Mitchell, J.E.; Lacey, C.A.; Harvest, C.K.; Li, L.; Nash, W.T.; Larson, H.N.; et al. Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 2022, 606, 960–967. [Google Scholar] [CrossRef]
- Kuechle, M.K.; Predd, H.M.; Fleckman, P.; Dale, B.A.; Presland, R.B. Caspase-14, a keratinocyte specific caspase: mRNA splice variants and expression pattern in embryonic and adult mouse. Cell Death Differ. 2001, 8, 868–870. [Google Scholar] [CrossRef]
- Pistritto, G.; Jost, M.; Srinivasula, S.M.; Baffa, R.; Poyet, J.-L.; Kari, C.; Lazebnik, Y.; Rodeck, U.; Alnemri, E.S.; Poyet, J.-L. Expression and transcriptional regulation of caspase-14 in simple and complex epithelia. Cell Death Differ. 2002, 9, 995–1006. [Google Scholar] [CrossRef]
- Rendl, M.; Ban, J.; Mrass, P.; Mayer, C.; Lengauer, B.; Eckhart, L.; Declerq, W.; Tschachler, E. Caspase-14 Expression by Epidermal Keratinocytes is Regulated by Retinoids in a Differentiation-associated Manner. J. Investig. Dermatol. 2002, 119, 1150–1155. [Google Scholar] [CrossRef]
- Galvan, V.; Roizman, B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 3931–3936. [Google Scholar] [CrossRef]
- Aubert, M.; O’Toole, J.; Blaho, J.A. Induction and Prevention of Apoptosis in Human HEp-2 Cells by Herpes Simplex Virus Type 1. J. Virol. 1999, 73, 10359–10370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanfilippo, C.M.; Blaho, J.A. ICP0 Gene Expression Is a Herpes Simplex Virus Type 1 Apoptotic Trigger. J. Virol. 2006, 80, 6810–6821. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Tamura, M.; Tanaka, S.; Fujii, T.; Aoki, A.; Komiyama, H.; Ezawa, K.; Sumiyama, K.; Sagai, T.; Shiroishi, T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 2007, 89, 618–629. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef]
- LaRock, D.L.; Johnson, A.F.; Wilde, S.; Sands, J.S.; Monteiro, M.P.; LaRock, C.N. Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 2022, 605, 527–531. [Google Scholar] [CrossRef]
- Saeki, N.; Kim, D.H.; Usui, T.; Aoyagi, K.; Tatsuta, T.; Aoki, K.; Yanagihara, K.; Tamura, M.; Mizushima, H.; Sakamoto, H.; et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-β-dependent apoptotic signalling. Oncogene 2007, 26, 6488–6498. [Google Scholar] [CrossRef]
- Shi, P.; Tang, A.; Xian, L.; Hou, S.; Zou, D.; Lv, Y.; Huang, Z.; Wang, Q.; Song, A.; Lin, Z.; et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem. J. 2015, 468, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Denecker, G.; Hoste, E.; Gilbert, B.; Hochepied, T.; Ovaere, P.; Lippens, S.; Broecke, C.V.D.; Van Damme, P.; D’Herde, K.; Hachem, J.-P.; et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat. Cell Biol. 2007, 9, 666–674. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered Cytokine Export and Apoptosis in Mice Deficient in Interleukin-1β Converting Enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M.; Macarak, E.J.; MacGregor, R.R.; Wolfe, J.; Kefalides, N.A. Virus Infection of Endothelial Cells. J. Infect Dis. 1981, 143, 266–273. [Google Scholar] [CrossRef]
- Purves, F.C.; Longnecker, R.M.; Leader, D.P.; Roizman, B. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 1987, 61, 2896–2901. [Google Scholar] [CrossRef]
- Brittle, E.E.; Wang, F.; Lubinski, J.M.; Bunte, R.M.; Friedman, H.M. A Replication-Competent, Neuronal Spread-Defective, Live Attenuated Herpes Simplex Virus Type 1 Vaccine. J. Virol. 2008, 82, 8431–8441. [Google Scholar] [CrossRef]
- Nagashunmugam, T.; Lubinski, J.; Wang, L.; Goldstein, L.T.; Weeks, B.S.; Sundaresan, P.; Kang, E.H.; Dubin, G.; Friedman, H.M. In Vivo Immune Evasion Mediated by the Herpes Simplex Virus Type 1 Immunoglobulin G Fc Receptor. J. Virol. 1998, 72, 5351–5359. [Google Scholar] [CrossRef]
- Yang, K.; Liang, Y.; Sun, Z.; Liu, L.; Liao, J.; Xu, H.; Zhu, M.; Fu, Y.-X.; Peng, H. T cell-derived lymphotoxin limits Th1 response during HSV-1 infection. Sci. Rep. 2018, 8, 17727. [Google Scholar] [CrossRef]
- Varanasi, S.K.; Rajasagi, N.K.; Jaggi, U.; Rouse, B.T. Role of IL-18 induced Amphiregulin expression on virus induced ocular lesions. Mucosal Immunol. 2018, 11, 1705–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakeman, F.D.; Whitley, R.J. Diagnosis of Herpes Simplex Encephalitis: Application of Polymerase Chain Reaction to Cerebrospinal Fluid from Brain-Biopsied Patients and Correlation with Disease. J. Infect. Dis. 1995, 171, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.T.; Short, G.A.; Irvine, A.R.; Duker, J.S.; Margolis, T.P. Acquired Immunodeficiency Syndrome—Associated Herpes Simplex Virus Retinitis: Clinical Description and Use of a Polymerase Chain Reaction—Based Assay as a Diagnostic Tool. Arch. Ophthalmol. 1996, 114, 834–840. [Google Scholar] [CrossRef]
- Bhullar, S.S.; Chandak, N.H.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F.; Kashyap, R.S. Determination of Viral Load by Quantitative Real-Time PCR in Herpes Simplex Encephalitis Patients. Intervirology 2013, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Orth, K.; Chinnaiyan, A.M.; Garg, M.; Froelich, C.J.; Dixit, V.M. The CED-3/ICE-like Protease Mch2 Is Activated during Apoptosis and Cleaves the Death Substrate Lamin A. J. Biol. Chem. 1996, 271, 16443–16446. [Google Scholar] [CrossRef]
- Hirata, H.; Takahashi, A.; Kobayashi, S.; Yonehara, S.; Sawai, H.; Okazaki, T.; Yamamoto, K.; Sasada, M. Caspases Are Activated in a Branched Protease Cascade and Control Distinct Downstream Processes in Fas-induced Apoptosis. J. Exp. Med. 1998, 187, 587–600. [Google Scholar] [CrossRef]
- Klaiman, G.; Champagne, N.; LeBlanc, A.C. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. BBA-Mol. Cell Res. 2009, 1793, 592–601. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Horowitz, P.M.; Patterson, K.R.; Guillozet-Bongaarts, A.L.; Reynolds, M.R.; Carroll, C.A.; Weintraub, S.T.; Bennett, D.A.; Cryns, V.L.; Berry, R.W.; Binder, L.I. Early N-Terminal Changes and Caspase-6 Cleavage of Tau in Alzheimer’s Disease. J. Neurosci. 2004, 24, 7895–7902. [Google Scholar] [CrossRef]
- Klaiman, G.; Petzke, T.L.; Hammond, J.; LeBlanc, A.C. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol. Cell. Proteom. MCP 2008, 7, 1541–1555. [Google Scholar] [CrossRef]
- LeBlanc, A.; Liu, H.; Goodyer, C.; Bergeron, C.; Hammond, J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999, 274, 23426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godefroy, N.; Foveau, B.; Albrecht, S.; Goodyer, C.G.; LeBlanc, A.C. Expression and Activation of Caspase-6 in Human Fetal and Adult Tissues. PLoS ONE 2013, 8, e79313. [Google Scholar]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Schoenmann, Z.; Assa-Kunik, E.; Tiomny, S.; Minis, A.; Haklai-Topper, L.; Arama, E.; Yaron, A. Axonal Degeneration Is Regulated by the Apoptotic Machinery or a NAD+-Sensitive Pathway in Insects and Mammals. J. Neurosci. 2010, 30, 6375–6386. [Google Scholar] [CrossRef]
- Cusack, C.L.; Swahari, V.; Henley, W.H.; Ramsey, J.M.; Deshmukh, M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat. Commun. 2013, 4, 1876. [Google Scholar] [CrossRef]
- Wang, X.-J.; Cao, Q.; Zhang, Y.; Su, X.-D. Activation and Regulation of Caspase-6 and Its Role in Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef]
- Low, L.K.; Cheng, H.-J. Axon pruning: An essential step underlying the developmental plasticity of neuronal connections. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1531–1544. [Google Scholar] [CrossRef]
- Taylor, M.P.; Enquist, L.W. Axonal spread of neuroinvasive viral infections. Trends Microbiol. 2015, 23, 283–288. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Tsunoda, I. Axonal degeneration as a self-destructive defense mechanism against neurotropic virus infection. Future Virol. 2008, 3, 579–593. [Google Scholar] [CrossRef]
- Milora, K.A.; Uppalapati, S.R.; Sanmiguel, J.C.; Zou, W.; Jensen, L.E. Interleukin-36β provides protection against HSV-1 infection, but does not modulate initiation of adaptive immune responses. Sci. Rep. 2017, 7, 5799. [Google Scholar] [CrossRef] [PubMed]
- Milora, K.A.; Miller, S.L.; Sanmiguel, J.C.; Jensen, L.E. Interleukin-1α released from HSV-1-infected keratinocytes acts as a functional alarmin in the skin. Nat. Commun. 2014, 5, 5230. [Google Scholar] [CrossRef] [Green Version]
- Khoury-Hanold, W.; Yordy, B.; Kong, P.; Kong, Y.; Ge, W.; Szigeti-Buck, K.; Ralevski, A.; Horvath, T.L.; Iwasaki, A. Viral Spread to Enteric Neurons Links Genital HSV-1 Infection to Toxic Megacolon and Lethality. Cell Host Microbe 2016, 19, 788–799. [Google Scholar] [CrossRef]
- Brun, P.; Qesari, M.; Marconi, P.C.R.; Kotsafti, A.; Porzionato, A.; Macchi, V.; Schwendener, R.A.; Scarpa, M.; Giron, M.C.; Palù, G.; et al. Herpes Simplex Virus Type 1 Infects Enteric Neurons and Triggers Gut Dysfunction via Macrophage Recruitment. Front. Cell. Infect. Microbiol. 2018, 8, 748. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.; Katze, M.G.; Gale, M.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Bala, U.; Tan, K.-L.; Ling, K.-H.; Cheah, P.-S. Harvesting the maximum length of sciatic nerve from adult mice: A step-by-step approach. BMC Res. Notes 2014, 7, 714. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, R.; Sant, C.V.; Roizman, B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. USA 1997, 94, 7891–7896. [Google Scholar] [CrossRef]
- Munger, J.; Roizman, B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10410–10415. [Google Scholar] [CrossRef]
- Hagglund, R.; Munger, J.; Poon, A.P.W.; Roizman, B. U S 3 Protein Kinase of Herpes Simplex Virus 1 Blocks Caspase 3 Activation Induced by the Products of U S 1.5 and U L 13 Genes and Modulates Expression of Transduced U S 1.5 Open Reading Frame in a Cell Type-Specific Manner. J. Virol. 2002, 76, 743–754. [Google Scholar] [CrossRef]
- Benetti, L.; Munger, J.; Roizman, B. The Herpes Simplex Virus 1 US3 Protein Kinase Blocks Caspase-Dependent Double Cleavage and Activation of the Proapoptotic Protein BAD. J. Virol. 2003, 77, 6567–6573. [Google Scholar] [CrossRef]
- Asano, S.; Honda, T.; Goshima, F.; Watanabe, D.; Miyake, Y.; Sugiura, Y.; Nishiyama, Y. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J. Gen. Virol. 1999, 80, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Asano, S.; Honda, T.; Goshima, F.; Nishiyama, Y.; Sugiura, Y. US3 protein kinase of herpes simplex virus protects primary afferent neurons from virus-induced apoptosis in ICR mice. Neurosci. Lett. 2000, 294, 105–108. [Google Scholar] [CrossRef]
- Mori, I.; Goshima, F.; Watanabe, D.; Ito, H.; Koide, N.; Yoshida, T.; Liu, B.; Kimura, Y.; Yokochi, T.; Nishiyama, Y. Herpes simplex virus US3 protein kinase regulates virus-induced apoptosis in olfactory and vomeronasal chemosensory neurons in vivo. Microbes Infect. 2006, 8, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, N.; Imai, T.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of herpes simplex virus 1 Us3 in viral neuroinvasiveness. Microbiol. Immunol. 2014, 58, 31–37. [Google Scholar] [CrossRef]
- Ghiasi, H.; Cai, S.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L. Perforin pathway is essential for protection of mice against lethal ocular HSV-1 challenge but not corneal scarring. Virus Res. 1999, 65, 97–101. [Google Scholar] [CrossRef]
- Chang, E.; Galle, L.; Maggs, D.; Estes, D.M.; Mitchell, W.J. Pathogenesis of Herpes Simplex Virus Type 1-Induced Corneal Inflammation in Perforin-Deficient Mice. J. Virol. 2000, 74, 11832–11840. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, S.Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, F.; Bhela, S.; Dogra, P.; Harvey, L.; Varanasi, S.K.; Jaggi, U.; Rouse, B.T. The inflammasome NLRP3 plays a protective role against a viral immunopathological lesion. J. Leukoc. Biol. 2016, 99, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strittmatter, G.E.; Sand, J.; Sauter, M.; Seyffert, M.; Steigerwald, R.; Fraefel, C.; Smola, S.; French, L.E.; Beer, H.-D. IFN-γ Primes Keratinocytes for HSV-1–Induced Inflammasome Activation. J. Investig. Dermatol. 2016, 136, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Lee, B.L.; Mirrashidi, K.M.; Stowe, I.B.; Kummerfeld, S.; Watanabe, C.; Haley, B.; Cuellar, T.L.; Reichelt, M.; Kayagaki, N. ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 2018, 8, 3788. [Google Scholar] [CrossRef]
- Orning, P.; Lien, E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J. Leukoc. Biol. 2021, 109, 121–141. [Google Scholar] [CrossRef]
- Kazem, S.; van der Meijden, E.; Struijk, L.; de Gruijl, F.R.; Feltkamp, M.C. Human papillomavirus 8 E6 disrupts terminal skin differentiation and prevents pro-Caspase-14 cleavage. Virus Res. 2012, 163, 609–616. [Google Scholar] [CrossRef]
- Yamamoto, M.; Miyai, M.; Matsumoto, Y.; Tsuboi, R.; Hibino, T. Kallikrein-related Peptidase-7 Regulates Caspase-14 Maturation during Keratinocyte Terminal Differentiation by Generating an Intermediate Form. J. Biol. Chem. 2012, 287, 32825–32834. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol. J. 2016, 13, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, M.; Blaho, J.A. The Herpes Simplex Virus Type 1 Regulatory Protein ICP27 Is Required for the Prevention of Apoptosis in Infected Human Cells. J. Virol. 1999, 73, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Jerome, K.R.; Fox, R.; Chen, Z.; Sears, A.E.; Lee, H.-Y.; Corey, L. Herpes Simplex Virus Inhibits Apoptosis through the Action of Two Genes, Us5 and Us3. J. Virol. 1999, 73, 8950–8957. [Google Scholar] [CrossRef] [PubMed]
- Jerome, K.R.; Chen, Z.; Lang, R.; Torres, M.R.; Hofmeister, J.; Smith, S.; Fox, R.; Froelich, C.J.; Corey, L. HSV and Glycoprotein J Inhibit Caspase Activation and Apoptosis Induced by Granzyme B or Fas. J. Immunol. 2001, 167, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the Herpes Simplex Virus Type 1 Latency-Associated Transcript That Protect Cells from Apoptosis In Vitro and Protect Neuronal Cells In Vivo. J. Virol. 2002, 76, 717–729. [Google Scholar] [CrossRef]
- Henderson, G.; Peng, W.; Jin, L.; Perng, G.-C.; Nesburn, A.; Wechsler, S.; Jones, C. Regulation of Caspase 8- and Caspase 9-Induced Apoptosis by the Herpes Simplex Virus Type 1 Latency-Associated Transcript. J. Neurovirol. 2002, 8, 103–111. [Google Scholar] [CrossRef]
- Wang, S.-L.; Zhao, G.; Zhu, W.; Dong, X.-M.; Liu, T.; Li, Y.-Y.; Song, W.-G.; Wang, Y.-Q. Herpes simplex virus-1 infection or Simian virus 40-mediated immortalization of corneal cells causes permanent translocation of NLRP3 to the nuclei. Int. J. Ophthalmol. 2014, 8, 46–51. [Google Scholar]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 2018, 23, 254–265.e7. [Google Scholar] [CrossRef]
- Fujioka, N.; Akazawa, R.; Ohashi, K.; Fujii, M.; Ikeda, M.; Kurimoto, M. Interleukin-18 Protects Mice against Acute Herpes Simplex Virus Type 1 Infection. J. Virol. 1999, 73, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Barr, D.P.; Belz, G.T.; Reading, P.C.; Wojtasiak, M.; Whitney, P.G.; Heath, W.R.; Carbone, F.R.; Brooks, A.G. A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. Eur. J. Immunol. 2007, 37, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Reading, P.C.; Whitney, P.; Barr, D.; Wojtasiak, M.; Mintern, J.; Waithman, J.; Brooks, A. IL-18, but not IL-12, Regulates NK Cell Activity following Intranasal Herpes Simplex Virus Type 1 Infection. J. Immunol. 2007, 179, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Sergerie, Y.; Rivest, S.; Boivin, G. Tumor Necrosis Factor-α and Interleukin-1β Play a Critical Role in the Resistance against Lethal Herpes Simplex Virus Encephalitis. J. Infect. Dis. 2007, 196, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Lucinda, N.; Figueiredo, M.M.; Pessoa, N.L.; Santos, B.S.; Lima, G.K.; Freitas, A.M.; Machado, A.M.; Kroon, E.G.; Antonelli, L.R.; Campos, M.A. Dendritic cells, macrophages, NK and CD8+ T lymphocytes play pivotal roles in controlling HSV-1 in the trigeminal ganglia by producing IL1-beta, iNOS and granzyme B. Virol. J. 2017, 14, 37. [Google Scholar] [CrossRef]
- Coulon, P.-G.; Dhanushkodi, N.; Prakash, S.; Srivastava, R.; Roy, S.; Alomari, N.I.; Nguyen, A.M.; Warsi, W.R.; Ye, C.; Carlos-Cruz, E.A.; et al. NLRP3, NLRP12, and IFI16 Inflammasomes Induction and Caspase-1 Activation Triggered by Virulent HSV-1 Strains Are Associated With Severe Corneal Inflammatory Herpetic Disease. Front. Immunol. 2019, 10, 1631. [Google Scholar] [CrossRef]
- Hayes, C.K.; Wilcox, D.R.; Yang, Y.; Coleman, G.K.; Brown, M.A.; Longnecker, R. ASC-dependent inflammasomes contribute to immunopathology and mortality in herpes simplex encephalitis. PLoS Pathog. 2021, 17, e1009285. [Google Scholar] [CrossRef]
- Hu, X.; Zeng, Q.; Xiao, J.; Qin, S.; Wang, Y.; Shan, T.; Hu, D.; Zhu, Y.; Liu, K.; Zheng, K.; et al. Herpes Simplex Virus 1 Induces Microglia Gasdermin D-Dependent Pyroptosis Through Activating the NLR Family Pyrin Domain Containing 3 Inflammasome. Front. Microbiol. 2022, 13, 838808. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Prochera, A.; Payne, L.; Smith, A.; Garlick, J.A.; Kagan, J.C. Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity 2021, 54, 1447–1462.e5. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Kovacs, S.B.; Jørgensen, I.; Larson, H.N.; Lazear, H.M.; Miao, E.A. Role of Caspases and Gasdermin A during HSV-1 Infection in Mice. Viruses 2022, 14, 2034. https://doi.org/10.3390/v14092034
Li L, Kovacs SB, Jørgensen I, Larson HN, Lazear HM, Miao EA. Role of Caspases and Gasdermin A during HSV-1 Infection in Mice. Viruses. 2022; 14(9):2034. https://doi.org/10.3390/v14092034
Chicago/Turabian StyleLi, Lupeng, Stephen B. Kovacs, Ine Jørgensen, Heather N. Larson, Helen M. Lazear, and Edward A. Miao. 2022. "Role of Caspases and Gasdermin A during HSV-1 Infection in Mice" Viruses 14, no. 9: 2034. https://doi.org/10.3390/v14092034
APA StyleLi, L., Kovacs, S. B., Jørgensen, I., Larson, H. N., Lazear, H. M., & Miao, E. A. (2022). Role of Caspases and Gasdermin A during HSV-1 Infection in Mice. Viruses, 14(9), 2034. https://doi.org/10.3390/v14092034