

Co-Infection and Cancer: Host–Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses

Abstract
:1. Background
2. HIV-1 and HTLV Coinfection
3. Dendritic Cells and HIV-1
3.1. Immunopathogenesis
3.2. Entry and Transmission
3.3. Immune Response
3.4. Cancer in HIV-1
3.5. Neuropathogenesis
3.6. Vaccine and Therapeutics
3.7. HIV/EBV Coinfection
4. Dendritic Cells and HTLV-1
4.1. Immunopathogenesis
4.2. Entry and Transmission
4.3. Immune Response
4.4. Cancer in HTLV-1
4.5. Neuropathogenesis
4.6. Vaccine and Therapeutics
4.7. HTLV/EBV Coinfection
5. DC Interaction with Other Human Oncogenic Viruses
5.1. EBV
5.2. HBV
5.3. HDV
5.4. HCV
5.5. HPV
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acquired Immunodeficiency Syndrome | AIDS |
| Adult T cell leukemia/lymphoma | ATLL |
| Alpha-fetoprotein | AFP |
| Antigen-presenting cells | APC |
| Antiretroviral therapy | ART |
| Chemokine receptor | CCR# |
| Cytotoxic lymphocytes | CTL |
| DC-specific intercellular adhesion molecule-3 grabbing non-integrin | DC-SIGN |
| Epstein–Barr virus | EBV |
| Glioblastoma | GBM |
| Head and neck squamous cell carcinomas | HNSCCs |
| Hepatitis B/C/D virus | HBV/HCV/HDV |
| Hepatocellular carcinoma | HCC |
| HIV-associated neurocognitive disorders | HAND |
| HTLV-1-associated myelopathy/tropical spastic paraparesis | HAM/TSP |
| Human immunodeficiency virus type 1 | HIV-1 |
| Human papillomavirus | HPV |
| Human T-lymphocytic virus type 1 | HTLV-1 |
| Interferon | IFN |
| Interleukin | IL |
| Long terminal repeat | LTR |
| Major histocompatibility complex | MHC |
| Myeloid dendritic cell and plasmacytoid dendritic cell | mDC and pDC |
| Natural killer cells | NK Cells |
| Pathogen-associated molecular patterns | PAMPs |
| Pattern recognition receptors | PRRs |
| People living with HIV | PLWH |
| Polymerase chain reaction | PCR |
| Protein negative factor | Nef |
| Reactive oxygen species | ROS |
| Regulatory T cells | T regs |
| Supramolecular activation complex | SMAC |
| Tumor necrosis factor | TNF |
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. 2), S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. How the immune system protects the host from infection. Microbes Infect. 2001, 3, 1167–1171. [Google Scholar] [CrossRef]
- Martin-Gayo, E.; Yu, X.G. Role of Dendritic Cells in Natural Immune Control of HIV-1 Infection. Front. Immunol. 2019, 10, 1306. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.K.; Platt, M.Y.; Eastham-Anderson, J.; Shin, J.S.; Mellman, I. MHC class II distribution in dendritic cells and B cells is determined by ubiquitin chain length. Proc. Natl. Acad. Sci. USA 2012, 109, 8820–8827. [Google Scholar] [CrossRef]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef]
- Munz, C. Antigen Processing for MHC Class II Presentation via Autophagy. Front. Immunol. 2012, 3, 9. [Google Scholar] [CrossRef]
- Jang, M.H.; Sougawa, N.; Tanaka, T.; Hirata, T.; Hiroi, T.; Tohya, K.; Guo, Z.; Umemoto, E.; Ebisuno, Y.; Yang, B.G.; et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J. Immunol. 2006, 176, 803–810. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Kondelkova, K.; Vokurkova, D.; Krejsek, J.; Borska, L.; Fiala, Z.; Ctirad, A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Med. 2010, 53, 73–77. [Google Scholar] [CrossRef]
- Castell-Rodríguez, A.; Piñón-Zárate, G.; Herrera-Enríquez, M.; Jarquín-Yáñez, K.; Medina-Solares, I. Dendritic cells: Location, function, and clinical implications. In Biology of Myelomonocytic Cells; IntechOpen: London, UK, 2017; pp. 21–50. [Google Scholar]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Sobenin, I.A.; Bobryshev, Y.V. Plasmacytoid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Front. Physiol. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology 2015, 220, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Bhardwaj, N. Dendritic cell dysregulation during HIV-1 infection. Immunol. Rev. 2013, 254, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Shimauchi, T.; Piguet, V. DC-T cell virological synapses and the skin: Novel perspectives in dermatology. Exp. Dermatol. 2015, 24, 1–4. [Google Scholar] [CrossRef]
- Araujo, A.Q. Neurological Aspects of HIV-1/HTLV-1 and HIV-1/HTLV-2 Coinfection. Pathogens 2020, 9, 250. [Google Scholar] [CrossRef]
- Zehender, G.; Meroni, L.; Varchetta, S.; De Maddalena, C.; Cavalli, B.; Gianotto, M.; Bosisio, A.B.; Colasante, C.; Rizzardini, G.; Moroni, M.; et al. Human T-lymphotropic virus type 2 (HTLV-2) provirus in circulating cells of the monocyte/macrophage lineage in patients dually infected with human immunodeficiency virus type 1 and HTLV-2 and having predominantly sensory polyneuropathy. J. Virol. 1998, 72, 7664–7668. [Google Scholar] [CrossRef]
- Moriuchi, H.; Moriuchi, M. In vitro induction of HIV-1 replication in resting CD4(+) T cells derived from individuals with undetectable plasma viremia upon stimulation with human T-cell leukemia virus type I. Virology 2000, 278, 514–519. [Google Scholar] [CrossRef]
- Siekevitz, M.; Josephs, S.F.; Dukovich, M.; Peffer, N.; Wong-Staal, F.; Greene, W.C. Activation of the HIV-1 LTR by T cell mitogens and the trans-activator protein of HTLV-I. Science 1987, 238, 1575–1578. [Google Scholar] [CrossRef]
- Zack, J.A.; Cann, A.J.; Lugo, J.P.; Chen, I.S. HIV-1 production from infected peripheral blood T cells after HTLV-I induced mitogenic stimulation. Science 1988, 240, 1026–1029. [Google Scholar] [CrossRef]
- De Rossi, A.; Saggioro, D.; Calabro, M.L.; Cenzato, R.; Chieco-Bianchi, L. Reciprocal activation of human T-lymphotropic viruses in HTLV-I-transformed cells superinfected with HIV-1. J. Acquir. Immune Defic. Syndr. 1991, 4, 380–385. [Google Scholar]
- Beilke, M.A.; Japa, S.; Vinson, D.G. HTLV-I and HTLV-II virus expression increase with HIV-1 coinfection. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bohnlein, E.; Siekevitz, M.; Ballard, D.W.; Lowenthal, J.W.; Rimsky, L.; Bogerd, H.; Hoffman, J.; Wano, Y.; Franza, B.R.; Greene, W.C. Stimulation of the human immunodeficiency virus type 1 enhancer by the human T-cell leukemia virus type I tax gene product involves the action of inducible cellular proteins. J. Virol. 1989, 63, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, N.; Cook, L.; Kagdi, H.; Basnayake, S.; Bangham, C.R.M.; Pozniak, A.L.; Taylor, G.P. Immune Compromise in HIV-1/HTLV-1 Coinfection with Paradoxical Resolution of CD4 Lymphocytosis during Antiretroviral Therapy: A Case Report. Medicine 2015, 94, e2275. [Google Scholar] [CrossRef] [PubMed]
- Pilotti, E.; Bianchi, M.V.; De Maria, A.; Bozzano, F.; Romanelli, M.G.; Bertazzoni, U.; Casoli, C. HTLV-1/-2 and HIV-1 co-infections: Retroviral interference on host immune status. Front. Microbiol. 2013, 4, 372. [Google Scholar] [CrossRef]
- Barrios, C.S.; Castillo, L.; Zhi, H.; Giam, C.Z.; Beilke, M.A. Human T cell leukaemia virus type 2 tax protein mediates CC-chemokine expression in peripheral blood mononuclear cells via the nuclear factor kappa B canonical pathway. Clin. Exp. Immunol. 2014, 175, 92–103. [Google Scholar] [CrossRef]
- Londhe, R.; Kulkarni, S. HTLV-2 Encoded Antisense Protein APH-2 Suppresses HIV-1 Replication. Viruses 2021, 13, 1432. [Google Scholar] [CrossRef]
- Ticona, E.; Huaman, M.A.; Yanque, O.; Zunt, J.R. HIV and HTLV-1 coinfection: The need to initiate antiretroviral therapy. J. Int. Assoc. Provid. AIDS Care 2013, 12, 373–374. [Google Scholar] [CrossRef]
- McLaren, P.J.; Fellay, J. HIV-1 and human genetic variation. Nat. Rev. Genet. 2021, 22, 645–657. [Google Scholar] [CrossRef]
- Friedland, G.H.; Klein, R.S. Transmission of the human immunodeficiency virus. N. Engl. J. Med. 1987, 317, 1125–1135. [Google Scholar] [CrossRef]
- Swanstrom, R.; Coffin, J. HIV-1 pathogenesis: The virus. Cold Spring Harb. Perspect. Med. 2012, 2, a007443. [Google Scholar] [CrossRef]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.H.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Y.; Khan, I.N.; Farman, M.; Al Karim, S.; Qadri, I.; Kamal, M.A.; Al Ghamdi, K.; Harakeh, S. HTLV-1 Associated Neurological Disorders. Curr. Top. Med. Chem. 2017, 17, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Smed-Sorensen, A.; Lore, K.; Vasudevan, J.; Louder, M.K.; Andersson, J.; Mascola, J.R.; Spetz, A.L.; Koup, R.A. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol. 2005, 79, 8861–8869. [Google Scholar] [CrossRef]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef]
- Coffin, J.M.; Varmus, H.E.; Bishop, J.M.; Essex, M.; Hardy, W.D., Jr.; Martin, G.S.; Rosenberg, N.E.; Scolnick, E.M.; Weinberg, R.A.; Vogt, P.K. Proposal for naming host cell-derived inserts in retrovirus genomes. J. Virol. 1981, 40, 953–957. [Google Scholar] [CrossRef]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- da Silva, R.C.; Segat, L.; Crovella, S. Role of DC-SIGN and L-SIGN receptors in HIV-1 vertical transmission. Hum. Immunol. 2011, 72, 305–311. [Google Scholar] [CrossRef]
- Bai, X.; Chen, J.D.; Yang, A.G.; Torti, F.; Chen, S.Y. Genetic co-inactivation of macrophage- and T-tropic HIV-1 chemokine coreceptors CCR-5 and CXCR-4 by intrakines. Gene Ther. 1998, 5, 984–994. [Google Scholar] [CrossRef]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef]
- Sehgal, M.; Khan, Z.K.; Talal, A.H.; Jain, P. Dendritic Cells in HIV-1 and HCV Infection: Can They Help Win the Battle? Virology 2013, 4, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.; Muñoz-Fernández, M.A. Dendritic Cells, the Double Agent in the War Against HIV-1. Front. Immunol. 2019, 10, 2485. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D. Dendritic Cells and HIV-1 Trans-Infection. Viruses 2010, 2, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Chang, E.; Sigal, A.; Baltimore, D. Dendritic cells efficiently transmit HIV to T Cells in a tenofovir and raltegravir insensitive manner. PLoS ONE 2018, 13, e0189945. [Google Scholar] [CrossRef]
- Vasiliver-Shamis, G.; Dustin, M.L.; Hioe, C.E. HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse. Viruses 2010, 2, 1239–1260. [Google Scholar] [CrossRef]
- Carrington, M.; Alter, G. Innate immune control of HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007070. [Google Scholar] [CrossRef]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef]
- Murugaiah, V.; Yasmin, H.; Pandit, H.; Ganguly, K.; Subedi, R.; Al-Mozaini, M.; Madan, T.; Kishore, U. Innate Immune Response against HIV-1. Adv. Exp. Med. Biol. 2021, 1313, 23–58. [Google Scholar] [CrossRef]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- van Montfort, T.; Eggink, D.; Boot, M.; Tuen, M.; Hioe, C.E.; Berkhout, B.; Sanders, R.W. HIV-1 N-glycan composition governs a balance between dendritic cell-mediated viral transmission and antigen presentation. J. Immunol. 2011, 187, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Upadhyay, C.; Hioe, C.E. HIV-1 Envelope Glycan Composition as a Key Determinant of Efficient Virus Transmission via DC-SIGN and Resistance to Inhibitory Lectins. iScience 2019, 21, 413–427. [Google Scholar] [CrossRef]
- Dieu, M.C.; Vanbervliet, B.; Vicari, A.; Bridon, J.M.; Oldham, E.; Aït-Yahia, S.; Brière, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Dave, R.S.; Jain, P.; Byrareddy, S.N. Follicular Dendritic Cells of Lymph Nodes as Human Immunodeficiency Virus/Simian Immunodeficiency Virus Reservoirs and Insights on Cervical Lymph Node. Front. Immunol. 2018, 9, 805. [Google Scholar] [CrossRef]
- Coulon, P.G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-Infected Dendritic Cells Present Endogenous MHC Class II-Restricted Antigens to HIV-Specific CD4+ T Cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef]
- Perreau, M.; Levy, Y.; Pantaleo, G. Immune response to HIV. Curr. Opin. HIV AIDS 2013, 8, 333–340. [Google Scholar] [CrossRef]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef]
- Chew, K.W.; Bhattacharya, D. Virologic and immunologic aspects of HIV-hepatitis C virus coinfection. AIDS 2016, 30, 2395–2404. [Google Scholar] [CrossRef]
- Sun, H.Y.; Sheng, W.H.; Tsai, M.S.; Lee, K.Y.; Chang, S.Y.; Hung, C.C. Hepatitis B virus coinfection in human immunodeficiency virus-infected patients: A review. World J. Gastroenterol. 2014, 20, 14598–14614. [Google Scholar] [CrossRef]
- Pinato, D.J.; Dalla Pria, A.; Sharma, R.; Bower, M. Hepatocellular carcinoma: An evolving challenge in viral hepatitis and HIV coinfection. AIDS 2017, 31, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; McGivern, D.R. Is hepatitis C virus carcinogenic? Gastroenterology 2012, 142, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Short, S.C. Management of glioblastoma multiforme in HIV patients: A case series and review of published studies. Clin. Oncol. (R. Coll. Radiol.) 2009, 21, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Pina-Oviedo, S. HIV disorders of the brain: Pathology and pathogenesis. Front. Biosci. 2006, 11, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Messam, C.A.; Major, E.O. Stages of restricted HIV-1 infection in astrocyte cultures derived from human fetal brain tissue. J. Neurovirol. 2000, 6 (Suppl. 1), S90–S94. [Google Scholar]
- Chiodi, F.; Fuerstenberg, S.; Gidlund, M.; Asjo, B.; Fenyo, E.M. Infection of brain-derived cells with the human immunodeficiency virus. J. Virol. 1987, 61, 1244–1247. [Google Scholar] [CrossRef]
- Dandachi, D.; Moron, F. Effects of HIV on the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1263, 45–54. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lien, K.; Mayer, W.; Herrera, R.; Rosbe, K.; Tugizov, S.M. HIV-1 proteins gp120 and tat induce the epithelial-mesenchymal transition in oral and genital mucosal epithelial cells. PLoS ONE 2019, 14, e0226343. [Google Scholar] [CrossRef]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress in Infection and Consequent Disease. Oxid. Med. Cell Longev. 2017, 2017, 3496043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayurova, E.; Jansons, J.; Skrastina, D.; Smirnova, O.; Mezale, D.; Kostyusheva, A.; Kostyushev, D.; Petkov, S.; Podschwadt, P.; Valuev-Elliston, V.; et al. HIV-1 Reverse Transcriptase Promotes Tumor Growth and Metastasis Formation via ROS-Dependent Upregulation of Twist. Oxid. Med. Cell Longev. 2019, 2019, 6016278. [Google Scholar] [CrossRef] [PubMed]
- Luckett, P.; Paul, R.H.; Navid, J.; Cooley, S.A.; Wisch, J.K.; Boerwinkle, A.H.; Tomov, D.; Ances, B.M. Deep Learning Analysis of Cerebral Blood Flow to Identify Cognitive Impairment and Frailty in Persons Living with HIV. J. Acquir. Immune Defic. Syndr. 2019, 82, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Boerwinkle, A.H.; Meeker, K.L.; Luckett, P.; Ances, B.M. Neuroimaging the Neuropathogenesis of HIV. Curr. HIV/AIDS Rep. 2021, 18, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B. HIV-associated neurocognitive disorder. Curr. Opin. Infect. Dis. 2017, 30, 117–122. [Google Scholar] [CrossRef]
- Valcour, V.; Chalermchai, T.; Sailasuta, N.; Marovich, M.; Lerdlum, S.; Suttichom, D.; Suwanwela, N.C.; Jagodzinski, L.; Michael, N.; Spudich, S.; et al. Central nervous system viral invasion and inflammation during acute HIV infection. J. Infect. Dis. 2012, 206, 275–282. [Google Scholar] [CrossRef]
- Hellmuth, J.; Valcour, V.; Spudich, S. CNS reservoirs for HIV: Implications for eradication. J. Virus Erad. 2015, 1, 67–71. [Google Scholar] [CrossRef]
- Kuhn, T.; Jin, Y.; Huang, C.; Kim, Y.; Nir, T.M.; Gullett, J.M.; Jones, J.D.; Sayegh, P.; Chung, C.; Dang, B.H.; et al. The joint effect of aging and HIV infection on microstructure of white matter bundles. Hum. Brain Mapp. 2019, 40, 4370–4380. [Google Scholar] [CrossRef]
- Gullett, J.M.; Lamb, D.G.; Porges, E.; Woods, A.J.; Rieke, J.; Thompson, P.; Jahanshad, N.; Nir, T.M.; Tashima, K.; Cohen, R.A. The Impact of Alcohol Use on Frontal White Matter in HIV. Alcohol. Clin. Exp. Res. 2018, 42, 1640–1649. [Google Scholar] [CrossRef]
- Valcour, V.; Sithinamsuwan, P.; Letendre, S.; Ances, B. Pathogenesis of HIV in the central nervous system. Curr. HIV/AIDS Rep. 2011, 8, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Groff, B.R.; Wiesman, A.I.; Rezich, M.T.; O’Neill, J.; Robertson, K.R.; Fox, H.S.; Swindells, S.; Wilson, T.W. Age-related visual dynamics in HIV-infected adults with cognitive impairment. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, R.H.; Cho, K.S.; Luckett, P.; Strain, J.F.; Belden, A.C.; Bolzenius, J.D.; Navid, J.; Garcia-Egan, P.M.; Cooley, S.A.; Wisch, J.K.; et al. Machine Learning Analysis Reveals Novel Neuroimaging and Clinical Signatures of Frailty in HIV. J. Acquir. Immune Defic. Syndr. 2020, 84, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Tomasi, D.; Yakupov, R.; Lozar, C.; Arnold, S.; Caparelli, E.; Ernst, T. Adaptation of the attention network in human immunodeficiency virus brain injury. Ann. Neurol. 2004, 56, 259–272. [Google Scholar] [CrossRef]
- Nickoloff-Bybel, E.A.; Festa, L.; Meucci, O.; Gaskill, P.J. Co-receptor signaling in the pathogenesis of neuroHIV. Retrovirology 2021, 18, 24. [Google Scholar] [CrossRef]
- Claing, A.; Laporte, S.A.; Caron, M.G.; Lefkowitz, R.J. Endocytosis of G protein-coupled receptors: Roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog. Neurobiol. 2002, 66, 61–79. [Google Scholar] [CrossRef]
- Zhang, J.; Barak, L.S.; Winkler, K.E.; Caron, M.G.; Ferguson, S.S. A central role for beta-arrestins and clathrin-coated vesicle-mediated endocytosis in beta2-adrenergic receptor resensitization. Differential regulation of receptor resensitization in two distinct cell types. J. Biol. Chem. 1997, 272, 27005–27014. [Google Scholar] [CrossRef]
- Gorska, A.M.; Eugenin, E.A. The Glutamate System as a Crucial Regulator of CNS Toxicity and Survival of HIV Reservoirs. Front. Cell Infect. Microbiol. 2020, 10, 261. [Google Scholar] [CrossRef]
- Sailasuta, N.; Shriner, K.; Ross, B. Evidence of reduced glutamate in the frontal lobe of HIV-seropositive patients. NMR Biomed. 2009, 22, 326–331. [Google Scholar] [CrossRef]
- Cohen, R.A.; Harezlak, J.; Gongvatana, A.; Buchthal, S.; Schifitto, G.; Clark, U.; Paul, R.; Taylor, M.; Thompson, P.; Tate, D.; et al. Cerebral metabolite abnormalities in human immunodeficiency virus are associated with cortical and subcortical volumes. J. Neurovirol. 2010, 16, 435–444. [Google Scholar] [CrossRef]
- Ernst, T.; Jiang, C.S.; Nakama, H.; Buchthal, S.; Chang, L. Lower brain glutamate is associated with cognitive deficits in HIV patients: A new mechanism for HIV-associated neurocognitive disorder. J. Magn. Reson. Imaging 2010, 32, 1045–1053. [Google Scholar] [CrossRef]
- Young, A.C.; Yiannoutsos, C.T.; Hegde, M.; Lee, E.; Peterson, J.; Walter, R.; Price, R.W.; Meyerhoff, D.J.; Spudich, S. Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology 2014, 83, 1592–1600. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.; Barker, P.B.; Skolasky, R.L.; Sacktor, N. 7T Brain MRS in HIV Infection: Correlation with Cognitive Impairment and Performance on Neuropsychological Tests. AJNR Am. J. Neuroradiol. 2018, 39, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Juge, L.; Lennon, M.J.; Gates, T.M.; Jones, S.P.; Lovelace, M.D.; Rae, C.D.; Johnson, T.P.; Nath, A.; Brew, B.J. HIV brain latency as measured by CSF BcL11b relates to disrupted brain cellular energy in virally suppressed HIV infection. AIDS 2019, 33, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Ferrarese, C.; Aliprandi, A.; Tremolizzo, L.; Stanzani, L.; De Micheli, A.; Dolara, A.; Frattola, L. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 2001, 57, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Misra, V.; Dutta, A.; Morgello, S.; Gabuzda, D. Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS 2014, 28, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Harezlak, J.; Bharti, A.; Mi, D.; Taylor, M.J.; Daar, E.S.; Schifitto, G.; Zhong, J.; Alger, J.R.; Brown, M.S.; et al. Plasma and Cerebrospinal Fluid Biomarkers Predict Cerebral Injury in HIV-Infected Individuals on Stable Combination Antiretroviral Therapy. J. Acquir. Immune Defic. Syndr. 2015, 69, 29–35. [Google Scholar] [CrossRef]
- Trickey, A.; May, M.T.; Schommers, P.; Tate, J.; Ingle, S.M.; Guest, J.L.; Gill, M.J.; Zangerle, R.; Saag, M.; Reiss, P.; et al. CD4:CD8 Ratio and CD8 Count as Prognostic Markers for Mortality in Human Immunodeficiency Virus-Infected Patients on Antiretroviral Therapy: The Antiretroviral Therapy Cohort Collaboration (ART-CC). Clin. Infect. Dis. 2017, 65, 959–966. [Google Scholar] [CrossRef]
- Roy, U.; Rodriguez, J.; Barber, P.; das Neves, J.; Sarmento, B.; Nair, M. The potential of HIV-1 nanotherapeutics: From in vitro studies to clinical trials. Nanomedicine 2015, 10, 3597–3609. [Google Scholar] [CrossRef]
- Rupp, R.; Rosenthal, S.L.; Stanberry, L.R. VivaGel (SPL7013 Gel): A candidate dendrimer—Microbicide for the prevention of HIV and HSV infection. Int. J. Nanomed. 2007, 2, 561–566. [Google Scholar]
- Gulick, R. HIV treatment 2020: What will it look like? J. Int. AIDS Soc. 2014, 17 (Suppl. 3), 19528. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.; Miller, V.; Jennings, S.R.; Wigdahl, B.; Krebs, F.C. The Evolution of Dendritic Cell Immunotherapy against HIV-1 Infection: Improvements and Outlook. J. Immunol. Res. 2020, 2020, 9470102. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Zanoni, I.; Feau, S.; Ricciardi-Castagnoli, P. Dendritic cell regulation of immune responses: A new role for interleukin 2 at the intersection of innate and adaptive immunity. EMBO J. 2003, 22, 2546–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamimi, C.; David, A.; Versmisse, P.; Weiss, L.; Bruel, T.; Zucman, D.; Appay, V.; Moris, A.; Ungeheuer, M.N.; Lascoux-Combe, C.; et al. Dendritic Cells from HIV Controllers Have Low Susceptibility to HIV-1 Infection In Vitro but High Capacity to Capture HIV-1 Particles. PLoS ONE 2016, 11, e0160251. [Google Scholar] [CrossRef]
- Smith, K.N.; Mailliard, R.B.; Larsen, B.B.; Wong, K.; Gupta, P.; Mullins, J.I.; Rinaldo, C.R. Dendritic cells restore CD8+ T cell reactivity to autologous HIV-1. J. Virol. 2014, 88, 9976–9990. [Google Scholar] [CrossRef]
- Herrera, C.; Harman, S.; Aldon, Y.; Rogers, P.; Armanasco, N.; Ziprin, P.; Stieh, D.; Nuttall, J.; Shattock, R.J. The entry inhibitor DS003 (BMS-599793): A BMS-806 analogue, provides superior activity as a pre-exposure prophylaxis candidate. AIDS 2021, 35, 1907–1917. [Google Scholar] [CrossRef]
- Lu, W.; Arraes, L.C.; Ferreira, W.T.; Andrieu, J.M. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat. Med. 2004, 10, 1359–1365. [Google Scholar] [CrossRef]
- Jiang, S.; Chan, C.N.; Rovira-Clave, X.; Chen, H.; Bai, Y.; Zhu, B.; McCaffrey, E.; Greenwald, N.F.; Liu, C.; Barlow, G.L.; et al. Combined protein and nucleic acid imaging reveals virus-dependent B cell and macrophage immunosuppression of tissue microenvironments. Immunity 2022, 55, 1118–1134 e1118. [Google Scholar] [CrossRef]
- Singh, A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat. Nanotechnol. 2021, 16, 16–24. [Google Scholar] [CrossRef]
- Gardner, M.R.; Kattenhorn, L.M.; Kondur, H.R.; von Schaewen, M.; Dorfman, T.; Chiang, J.J.; Haworth, K.G.; Decker, J.M.; Alpert, M.D.; Bailey, C.C.; et al. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature 2015, 519, 87–91. [Google Scholar] [CrossRef]
- Dunleavy, K.; Wilson, W.H. How I treat HIV-associated lymphoma. Blood 2012, 119, 3245–3255. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Rizk, M.; Teklezghi, A.; Spagnuolo, R.A.; Pagano, J.S.; Wahl, A. HIV co-infection augments EBV-induced tumorigenesis in vivo. Front. Virol. 2022, 2, 861628. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.Y.; Kaul, M. Beneficial and Adverse Effects of cART Affect Neurocognitive Function in HIV-1 Infection: Balancing Viral Suppression against Neuronal Stress and Injury. J. Neuroimmune Pharmacol. 2021, 16, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, D.; Bromberg, J.E.C. Primary CNS lymphoma in HIV infection. Handb. Clin. Neurol. 2018, 152, 177–186. [Google Scholar] [CrossRef]
- Lang, F.; Pei, Y.; Lamplugh, Z.L.; Robertson, E.S. Molecular Biology of EBV in Relationship to HIV/AIDS-Associated Oncogenesis. Cancer Treat. Res. 2019, 177, 81–103. [Google Scholar] [CrossRef]
- Zhang, N.; Zuo, Y.; Jiang, L.; Peng, Y.; Huang, X.; Zuo, L. Epstein-Barr Virus and Neurological Diseases. Front. Mol. Biosci. 2021, 8, 816098. [Google Scholar] [CrossRef]
- Byrne, C.M.; Johnston, C.; Orem, J.; Okuku, F.; Huang, M.L.; Rahman, H.; Wald, A.; Corey, L.; Schiffer, J.T.; Casper, C.; et al. Examining the dynamics of Epstein-Barr virus shedding in the tonsils and the impact of HIV-1 coinfection on daily saliva viral loads. PLoS Comput. Biol. 2021, 17, e1009072. [Google Scholar] [CrossRef]
- Hajtovic, S.; Liu, C.; Diefenbach, C.M.; Placantonakis, D.G. Epstein-Barr Virus-Positive Primary Central Nervous System Lymphoma in a 40-Year-Old Immunocompetent Patient. Cureus 2021, 13, e12754. [Google Scholar] [CrossRef]
- Hoshino, H. Cellular Factors Involved in HTLV-1 Entry and Pathogenicit. Front Microbiol. 2012, 3, 222. [Google Scholar]
- World Health Organization. Human T-Lymphotropic Virus Type 1: Technical Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef]
- Goncalves, D.U.; Proietti, F.A.; Ribas, J.G.; Araujo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef]
- Fuzii, H.T.; da Silva Dias, G.A.; de Barros, R.J.; Falcao, L.F.; Quaresma, J.A. Immunopathogenesis of HTLV-1-assoaciated myelopathy/tropical spastic paraparesis (HAM/TSP). Life Sci. 2014, 104, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Thoma-Kress, A.K. Molecular Mechanisms of HTLV-1 Cell-to-Cell Transmission. Viruses 2016, 8, 74. [Google Scholar] [CrossRef]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef]
- Alais, S.; Mahieux, R.; Dutartre, H. Viral Source-Independent High Susceptibility of Dendritic Cells to Human T-Cell Leukemia Virus Type 1 Infection Compared to That of T Lymphocytes. J. Virol. 2015, 89, 10580–10590. [Google Scholar] [CrossRef] [PubMed]
- Kampani, K.; Quann, K.; Ahuja, J.; Wigdahl, B.; Khan, Z.K.; Jain, P. A novel high throughput quantum dot-based fluorescence assay for quantitation of virus binding and attachment. J. Virol. Methods 2007, 141, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Maldonado, J.O.; Mueller, J.D.; Zhang, W.; Mansky, L.M. Molecular Studies of HTLV-1 Replication: An Update. Viruses 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Manuel, S.L.; Khan, Z.K.; Ahuja, J.; Quann, K.; Wigdahl, B. DC-SIGN mediates cell-free infection and transmission of human T-cell lymphotropic virus type 1 by dendritic cells. J. Virol. 2009, 83, 10908–10921. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Negi, V.S.; Mukherjee, S.; Tanaka, Y.; Orth, K.; Taylor, G.P.; Bangham, C.R. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 2009, 114, 1016–1025. [Google Scholar] [CrossRef]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef]
- Eusebio-Ponce, E.; Anguita, E.; Paulino-Ramirez, R.; Candel, F.J. HTLV-1 infection: An emerging risk. Pathogenesis, epidemiology, diagnosis and associated diseases. Rev. Esp Quimioter. 2019, 32, 485–496. [Google Scholar]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef] [PubMed]
- Malbec, M.; Roesch, F.; Schwartz, O. A new role for the HTLV-1 p8 protein: Increasing intercellular conduits and viral cell-to-cell transmission. Viruses 2011, 3, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Journo, C.; Mahieux, R. HTLV-1 and innate immunity. Viruses 2011, 3, 1374–1394. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Shin, H.W.; Lee, J.M.; Lee, H.W.; Kim, E.C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediators Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizkallah, G.; Alais, S.; Futsch, N.; Tanaka, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Dendritic cell maturation, but not type I interferon exposure, restricts infection by HTLV-1, and viral transmission to T-cells. PLoS Pathog. 2017, 13, e1006353. [Google Scholar] [CrossRef]
- Norris, P.J.; Hirschkorn, D.F.; DeVita, D.A.; Lee, T.H.; Murphy, E.L. Human T cell leukemia virus type 1 infection drives spontaneous proliferation of natural killer cells. Virulence 2010, 1, 19–28. [Google Scholar] [CrossRef]
- Rafatpanah, H.; Farid Hosseini, R.; Pourseyed, S.H. The Impact of Immune Response on HTLV-I in HTLV-I-Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP). Iran. J. Basic Med. Sci. 2013, 16, 235–241. [Google Scholar]
- Biddison, W.E.; Kubota, R.; Kawanishi, T.; Taub, D.D.; Cruikshank, W.W.; Center, D.M.; Connor, E.W.; Utz, U.; Jacobson, S. Human T cell leukemia virus type I (HTLV-I)-specific CD8+ CTL clones from patients with HTLV-I-associated neurologic disease secrete proinflammatory cytokines, chemokines, and matrix metalloproteinase. J. Immunol. 1997, 159, 2018–2025. [Google Scholar]
- Goon, P.K.; Hanon, E.; Igakura, T.; Tanaka, Y.; Weber, J.N.; Taylor, G.P.; Bangham, C.R. High frequencies of Th1-type CD4(+) T cells specific to HTLV-1 Env and Tax proteins in patients with HTLV-1-associated myelopathy/tropical spastic paraparesis. Blood 2002, 99, 3335–3341. [Google Scholar] [CrossRef]
- Satou, Y.; Utsunomiya, A.; Tanabe, J.; Nakagawa, M.; Nosaka, K.; Matsuoka, M. HTLV-1 modulates the frequency and phenotype of FoxP3+CD4+ T cells in virus-infected individuals. Retrovirology 2012, 9, 46. [Google Scholar] [CrossRef]
- Iwanaga, M.; Watanabe, T.; Utsunomiya, A.; Okayama, A.; Uchimaru, K.; Koh, K.R.; Ogata, M.; Kikuchi, H.; Sagara, Y.; Uozumi, K.; et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: A nationwide prospective study in Japan. Blood 2010, 116, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T. Adult T-cell leukemia: Molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 2017, 129, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Weinstock, D.M. Clinicogenetic risk modeling in ATL. Blood 2018, 131, 159–160. [Google Scholar] [CrossRef]
- Bangham, C.R.; Ratner, L. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr. Opin. Virol. 2015, 14, 93–100. [Google Scholar] [CrossRef]
- Grossman, W.J.; Kimata, J.T.; Wong, F.H.; Zutter, M.; Ley, T.J.; Ratner, L. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc. Natl. Acad. Sci. USA 1995, 92, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Sawa, H.; Lewis, M.J.; Orba, Y.; Sheehy, N.; Yamamoto, Y.; Ichinohe, T.; Tsunetsugu-Yokota, Y.; Katano, H.; Takahashi, H.; et al. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat. Med. 2006, 12, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Ohsugi, T. A transgenic mouse model of human T cell leukemia virus type 1-associated diseases. Front. Microbiol. 2013, 4, 49. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: Viral infectivity, Tax, HBZ and therapy. Oncogene 2011, 30, 1379–1389. [Google Scholar] [CrossRef]
- Choi, Y.B.; Harhaj, E.W. HTLV-1 tax stabilizes MCL-1 via TRAF6-dependent K63-linked polyubiquitination to promote cell survival and transformation. PLoS Pathog. 2014, 10, e1004458. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- Baydoun, H.H.; Bai, X.T.; Shelton, S.; Nicot, C. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS ONE 2012, 7, e42226. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Shimoyama, M.; Tobinai, K.; Ito, M.; Ito, S.; Ikeda, S.; Tajima, K.; Shimotohno, K.; Sugimura, T. Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 5620–5624. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Green, P.L. The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology 2009, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.S.; Villaudy, J.; Gazzolo, L.; Castellazzi, M.; Mesnard, J.M.; Duc Dodon, M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 2007, 4, 92. [Google Scholar] [CrossRef]
- Abolbashari, S.; Ghayour-Mobarhan, M.; Ebrahimi, M.; Meshkat, Z. The role of human T-lymphotropic virus (HTLV) in cardiovascular diseases: A review of literature. ARYA Atheroscler. 2018, 14, 183–187. [Google Scholar] [CrossRef]
- Quaresma, J.A.; Yoshikawa, G.T.; Koyama, R.V.; Dias, G.A.; Fujihara, S.; Fuzii, H.T. HTLV-1, Immune Response and Autoimmunity. Viruses 2015, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Izumo, S. Neuropathology of HTLV-1-associated myelopathy (HAM/TSP): The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology 2010, 30, 480–485. [Google Scholar] [CrossRef]
- Enose-Akahata, Y.; Jacobson, S. Immunovirological markers in HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 2019, 16, 35. [Google Scholar] [CrossRef]
- Oh, U.; Jacobson, S. Treatment of HTLV-I-associated myelopathy/tropical spastic paraparesis: Toward rational targeted therapy. Neurol. Clin. 2008, 26, 781–797, ix–x. [Google Scholar] [CrossRef]
- Manuel, S.L.; Sehgal, M.; Khan, Z.K.; Goedert, J.J.; Betts, M.R.; Jain, P. An altered maturation and adhesion phenotype of dendritic cells in diseased individuals compared to asymptomatic carriers of human T cell leukemia virus type 1. AIDS Res. Hum. Retrovir. 2013, 29, 1273–1285. [Google Scholar] [CrossRef]
- Jacobson, S.; Shida, H.; McFarlin, D.E.; Fauci, A.S.; Koenig, S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature 1990, 348, 245–248. [Google Scholar] [CrossRef]
- Manuel, S.L.; Rahman, S.; Wigdahl, B.; Khan, Z.K.; Jain, P. Dendritic cells in autoimmune diseases and neuroinflammatory disorders. Front. Biosci. 2007, 12, 4315–4335. [Google Scholar] [CrossRef]
- Sagar, D.; Masih, S.; Schell, T.; Jacobson, S.; Comber, J.D.; Philip, R.; Wigdahl, B.; Jain, P.; Khan, Z.K. In vivo immunogenicity of Tax(11-19) epitope in HLA-A2/DTR transgenic mice: Implication for dendritic cell-based anti-HTLV-1 vaccine. Vaccine 2014, 32, 3274–3284. [Google Scholar] [CrossRef]
- Kannagi, M.; Hasegawa, A.; Nagano, Y.; Iino, T.; Okamura, J.; Suehiro, Y. Maintenance of long remission in adult T-cell leukemia by Tax-targeted vaccine: A hope for disease-preventive therapy. Cancer Sci. 2019, 110, 849–857. [Google Scholar] [CrossRef]
- Khaddour, K.; Hana, C.K.; Mewawalla, P. Hematopoietic Stem Cell Transplantation. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Marino-Merlo, F.; Balestrieri, E.; Matteucci, C.; Mastino, A.; Grelli, S.; Macchi, B. Antiretroviral Therapy in HTLV-1 Infection: An Updated Overview. Pathogens 2020, 9, 342. [Google Scholar] [CrossRef]
- Matsushita, S.; Mitsuya, H.; Reitz, M.S.; Broder, S. Pharmacological inhibition of in vitro infectivity of human T lymphotropic virus type I. J. Clin. Invest. 1987, 80, 394–400. [Google Scholar] [CrossRef]
- Isono, T.; Ogawa, K.; Seto, A. Antiviral effect of zidovudine in the experimental model of adult T cell leukemia in rabbits. Leuk Res. 1990, 14, 841–847. [Google Scholar] [CrossRef]
- Macchi, B.; Faraoni, I.; Zhang, J.; Grelli, S.; Favalli, C.; Mastino, A.; Bonmassar, E. AZT inhibits the transmission of human T cell leukaemia/lymphoma virus type I to adult peripheral blood mononuclear cells in vitro. J. Gen. Virol. 1997, 78 Pt 5, 1007–1016. [Google Scholar] [CrossRef]
- Gout, O.; Gessain, A.; Iba-Zizen, M.; Kouzan, S.; Bolgert, F.; de The, G.; Lyon-Caen, O. The effect of zidovudine on chronic myelopathy associated with HTLV-1. J. Neurol. 1991, 238, 108–109. [Google Scholar] [CrossRef]
- Afonso, P.V.; Mekaouche, M.; Mortreux, F.; Toulza, F.; Moriceau, A.; Wattel, E.; Gessain, A.; Bangham, C.R.; Dubreuil, G.; Plumelle, Y.; et al. Highly active antiretroviral treatment against STLV-1 infection combining reverse transcriptase and HDAC inhibitors. Blood 2010, 116, 3802–3808. [Google Scholar] [CrossRef]
- Hermine, O.; Bouscary, D.; Gessain, A.; Turlure, P.; Leblond, V.; Franck, N.; Buzyn-Veil, A.; Rio, B.; Macintyre, E.; Dreyfus, F.; et al. Brief report: Treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N. Engl. J. Med. 1995, 332, 1749–1751. [Google Scholar] [CrossRef]
- Ando, S.; Hasegawa, A.; Murakami, Y.; Zeng, N.; Takatsuka, N.; Maeda, Y.; Masuda, T.; Suehiro, Y.; Kannagi, M. HTLV-1 Tax-Specific CTL Epitope-Pulsed Dendritic Cell Therapy Reduces Proviral Load in Infected Rats with Immune Tolerance against Tax. J. Immunol. 2017, 198, 1210–1219. [Google Scholar] [CrossRef]
- Sasada, A.; Takaori-Kondo, A.; Shirakawa, K.; Kobayashi, M.; Abudu, A.; Hishizawa, M.; Imada, K.; Tanaka, Y.; Uchiyama, T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2005, 2, 32. [Google Scholar] [CrossRef]
- Izumi, T.; Shirakawa, K.; Takaori-Kondo, A. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev. Med. Chem. 2008, 8, 231–238. [Google Scholar] [CrossRef]
- Ceccaldi, P.E.; Delebecque, F.; Prevost, M.C.; Moris, A.; Abastado, J.P.; Gessain, A.; Schwartz, O.; Ozden, S. DC-SIGN facilitates fusion of dendritic cells with human T-cell leukemia virus type 1-infected cells. J. Virol. 2006, 80, 4771–4780. [Google Scholar] [CrossRef]
- Yao, J.; Tanaka, M.; Takenouchi, N.; Ren, Y.; Lee, S.I.; Fujisawa, J.I. Induction of APOBEC3B cytidine deaminase in HTLV-1-infected humanized mice. Exp. Ther. Med. 2019, 17, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Beltran, B.; Salas, R.; Quinones, P.; Morales, D.; Hurtado, F.; Cotrina, E.; Riva, L.; Castillo, J. EBV-positive diffuse large B-cell lymphoma in a human T-lymphotropic virus type 1 carrier. Infect. Agent Cancer 2009, 4, 10. [Google Scholar] [CrossRef]
- Pagano, J.S.; Kenney, S.; Markovitz, D.; Kamine, J. Epstein-Barr virus and interactions with human retroviruses. J. Virol. Methods 1988, 21, 229–239. [Google Scholar] [CrossRef]
- Nishida, Y.; Hyakuna, Y.; Fujisawa, K.; Yamano, Y.; Ohshima, K.; Higuchi, M. Composite lymphoma consisting of adult T-cell leukemia-lymphoma and diffuse large B-cell lymphoma. Rinsho Ketsueki 2013, 54, 2062–2067. [Google Scholar]
- Ueda, S.; Maeda, Y.; Yamaguchi, T.; Hanamoto, H.; Hijikata, Y.; Tanaka, M.; Takai, S.; Hirase, C.; Morita, Y.; Kanamaru, A. Influence of Epstein-Barr virus infection in adult T-cell leukemia. Hematology 2008, 13, 154–162. [Google Scholar] [CrossRef]
- Sakamoto, H.; Imaizumi, Y.; Niino, D.; Takeuchi, M.; Matsui, K.; Horai, M.; Sato, S.; Akazawa, Y.; Ando, K.; Sawayama, Y.; et al. Composite adult T-cell leukemia/lymphoma and Epstein-Barr virus-positive diffuse large B-cell lymphoma. Rinsho Ketsueki 2020, 61, 305–311. [Google Scholar] [CrossRef]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K.; et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019, 29, 2321–2337.e2327. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef]
- Pagano, J.S. Epstein-Barr virus: The first human tumor virus and its role in cancer. Proc. Assoc. Am. Physicians 1999, 111, 573–580. [Google Scholar] [CrossRef]
- Roberts, M.L.; Luxembourg, A.T.; Cooper, N.R. Epstein-Barr virus binding to CD21, the virus receptor, activates resting B cells via an intracellular pathway that is linked to B cell infection. J. Gen. Virol. 1996, 77 Pt 12, 3077–3085. [Google Scholar] [CrossRef]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr Virus Infection in the Development of Neurological Disorders. Drug Discov. Today Dis. Models 2020, 32, 35–52. [Google Scholar] [CrossRef]
- Munz, C. Dendritic cells during Epstein Barr virus infection. Front. Microbiol. 2014, 5, 308. [Google Scholar] [CrossRef]
- Lotz, M.; Tsoukas, C.D.; Fong, S.; Carson, D.A.; Vaughan, J.H. Regulation of Epstein-Barr virus infection by recombinant interferons. Selected sensitivity to interferon-gamma. Eur. J. Immunol. 1985, 15, 520–525. [Google Scholar] [CrossRef]
- Lim, W.H.; Kireta, S.; Russ, G.R.; Coates, P.T. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 2007, 109, 1043–1050. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Pack, M.; Thomas, D.; Paludan, C.; Schmid, D.; Strowig, T.; Bougras, G.; Muller, W.A.; Moretta, L.; Munz, C. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc. Natl. Acad. Sci. USA 2004, 101, 16606–16611. [Google Scholar] [CrossRef]
- Strowig, T.; Brilot, F.; Arrey, F.; Bougras, G.; Thomas, D.; Muller, W.A.; Munz, C. Tonsilar NK cells restrict B cell transformation by the Epstein-Barr virus via IFN-gamma. PLoS Pathog. 2008, 4, e27. [Google Scholar] [CrossRef]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef]
- Bickham, K.; Goodman, K.; Paludan, C.; Nikiforow, S.; Tsang, M.L.; Steinman, R.M.; Munz, C. Dendritic cells initiate immune control of epstein-barr virus transformation of B lymphocytes in vitro. J. Exp. Med. 2003, 198, 1653–1663. [Google Scholar] [CrossRef]
- Shih, C.; Yang, C.C.; Choijilsuren, G.; Chang, C.H.; Liou, A.T. Hepatitis B Virus. Trends Microbiol. 2018, 26, 386–387. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Tsuei, D.J.; Lee, P.H.; Peng, H.Y.; Lu, H.L.; Su, D.S.; Jeng, Y.M.; Hsu, H.C.; Hsu, S.H.; Wu, J.F.; Ni, Y.H.; et al. Male germ cell-specific RNA binding protein RBMY: A new oncogene explaining male predominance in liver cancer. PLoS ONE 2011, 6, e26948. [Google Scholar] [CrossRef]
- Dane, D.S.; Cameron, C.H.; Briggs, M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1970, 1, 695–698. [Google Scholar] [CrossRef]
- Yamada, G.; Sakamoto, Y.; Mizuno, M.; Nishihara, T.; Kobayashi, T.; Takahashi, T.; Nagashima, H. Electron and immunoelectron microscopic study of Dane particle formation in chronic hepatitis B virus infection. Gastroenterology 1982, 83, 348–356. [Google Scholar] [CrossRef]
- Venkatakrishnan, B.; Zlotnick, A. The Structural Biology of Hepatitis B Virus: Form and Function. Annu. Rev. Virol. 2016, 3, 429–451. [Google Scholar] [CrossRef]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef]
- Saxena, N.K.; Sharma, D.; Ding, X.; Lin, S.; Marra, F.; Merlin, D.; Anania, F.A. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Wang, Y.; Wang, P.; Chen, K.; Wang, M.; Zeng, H.; Lu, J.; Song, Q.; Diplas, B.H.; Tan, D.; et al. Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy. Proc. Natl. Acad. Sci. USA 2019, 116, 6308–6312. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Fan, J.; Yang, X.R.; Tan, Y.; Zhao, W.; Xu, Y.; Wang, N.; Niu, Y.; Wu, Z.; Zhou, J.; et al. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: A large-scale, multicentre study. Lancet Oncol. 2012, 13, 817–826. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Patel, E.U.; Thio, C.L.; Boon, D.; Thomas, D.L.; Tobian, A.A.R. Prevalence of Hepatitis B and Hepatitis D Virus Infections in the United States, 2011–2016. Clin. Infect. Dis. 2019, 69, 709–712. [Google Scholar] [CrossRef]
- Taylor, J.M. Infection by Hepatitis Delta Virus. Viruses 2020, 12, 648. [Google Scholar] [CrossRef]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Xu, X.; Xu, Y.; Li, C.; Xie, F.; Guo, M.; Liu, Z.; Liu, X. OGDHL ameliorates cognitive impairment and Alzheimer’s disease-like pathology via activating Wnt/beta-catenin signaling in Alzheimer’s disease mice. Behav. Brain Res. 2022, 418, 113673. [Google Scholar] [CrossRef]
- Riccitelli, N.; Luptak, A. HDV family of self-cleaving ribozymes. Prog. Mol. Biol. Transl. Sci. 2013, 120, 123–171. [Google Scholar] [CrossRef]
- Zheng, L.; Falschlunger, C.; Huang, K.; Mairhofer, E.; Yuan, S.; Wang, J.; Patel, D.J.; Micura, R.; Ren, A. Hatchet ribozyme structure and implications for cleavage mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 10783–10791. [Google Scholar] [CrossRef]
- McCormick, F. Cancer gene therapy: Fringe or cutting edge? Nat. Rev. Cancer 2001, 1, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Lortat-Jacob, H.; de Lacroix de Lavalette, A.; Staropoli, I.; Foung, S.; Amara, A.; Houles, C.; Fieschi, F.; Schwartz, O.; Virelizier, J.L.; et al. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003, 278, 20358–20366. [Google Scholar] [CrossRef] [PubMed]
- Sagan, S.M.; Sarnow, P. Plasmacytoid dendritic cells as guardians in hepatitis C virus-infected liver. Proc. Natl. Acad. Sci. USA 2010, 107, 7625–7626. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Levine, B. Autophagy in cellular growth control. FEBS Lett. 2010, 584, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Raposo, G. Exosomes--vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Moriya, O.; Matsui, M.; Osorio, M.; Miyazawa, H.; Rice, C.M.; Feinstone, S.M.; Leppla, S.H.; Keith, J.M.; Akatsuka, T. Induction of hepatitis C virus-specific cytotoxic T lymphocytes in mice by immunization with dendritic cells treated with an anthrax toxin fusion protein. Vaccine 2001, 20, 789–796. [Google Scholar] [CrossRef]
- Matsui, M.; Moriya, O.; Abdel-Aziz, N.; Matsuura, Y.; Miyamura, T.; Akatsuka, T. Induction of hepatitis C virus-specific cytotoxic T lymphocytes in mice by immunization with dendritic cells transduced with replication-defective recombinant adenovirus. Vaccine 2002, 21, 211–220. [Google Scholar] [CrossRef]
- Zabaleta, A.; Llopiz, D.; Arribillaga, L.; Silva, L.; Riezu-Boj, J.I.; Lasarte, J.J.; Borras-Cuesta, F.; Prieto, J.; Sarobe, P. Vaccination against hepatitis C virus with dendritic cells transduced with an adenovirus encoding NS3 protein. Mol. Ther. 2008, 16, 210–217. [Google Scholar] [CrossRef]
- Jirmo, A.C.; Koya, R.C.; Sundarasetty, B.S.; Pincha, M.; Yu, G.Y.; Lai, M.; Bakshi, R.; Schlaphoff, V.; Grabowski, J.; Behrens, G.; et al. Monocytes transduced with lentiviral vectors expressing hepatitis C virus non-structural proteins and differentiated into dendritic cells stimulate multi-antigenic CD8(+) T cell responses. Vaccine 2010, 28, 922–933. [Google Scholar] [CrossRef]
- Gehring, S.; Gregory, S.H.; Wintermeyer, P.; Aloman, C.; Wands, J.R. Generation of immune responses against hepatitis C virus by dendritic cells containing NS5 protein-coated microparticles. Clin. Vaccine Immunol. 2009, 16, 163–171. [Google Scholar] [CrossRef]
- Wintermeyer, P.; Gehring, S.; Eken, A.; Wands, J.R. Generation of cellular immune responses to HCV NS5 protein through in vivo activation of dendritic cells. J. Viral Hepat. 2010, 17, 705–713. [Google Scholar] [CrossRef]
- Barth, H.; Ulsenheimer, A.; Pape, G.R.; Diepolder, H.M.; Hoffmann, M.; Neumann-Haefelin, C.; Thimme, R.; Henneke, P.; Klein, R.; Paranhos-Baccala, G.; et al. Uptake and presentation of hepatitis C virus-like particles by human dendritic cells. Blood 2005, 105, 3605–3614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Yao, Z.; Moorman, J.P.; Jia, Z. Dendritic cell-based immunity and vaccination against hepatitis C virus infection. Immunology 2012, 136, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Pantel, A.; Cheong, C.; Dandamudi, D.; Shrestha, E.; Mehandru, S.; Brane, L.; Ruane, D.; Teixeira, A.; Bozzacco, L.; Steinman, R.M.; et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T-cell immunity in vivo. Eur. J. Immunol. 2012, 42, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef]
- Song, X.T.; Evel-Kabler, K.; Shen, L.; Rollins, L.; Huang, X.F.; Chen, S.Y. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat. Med. 2008, 14, 258–265. [Google Scholar] [CrossRef]
- Van Renne, N.; Roca Suarez, A.A.; Duong, F.H.T.; Gondeau, C.; Calabrese, D.; Fontaine, N.; Ababsa, A.; Bandiera, S.; Croonenborghs, T.; Pochet, N.; et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 2018, 67, 953–962. [Google Scholar] [CrossRef]
- Kawamura, H.; Govindarajan, S.; Aswad, F.; Machida, K.; Lai, M.M.; Sung, V.M.; Dennert, G. HCV core expression in hepatocytes protects against autoimmune liver injury and promotes liver regeneration in mice. Hepatology 2006, 44, 936–944. [Google Scholar] [CrossRef]
- Pynnonen, M.A.; Gillespie, M.B.; Roman, B.; Rosenfeld, R.M.; Tunkel, D.E.; Bontempo, L.; Brook, I.; Chick, D.A.; Colandrea, M.; Finestone, S.A.; et al. Clinical Practice Guideline: Evaluation of the Neck Mass in Adults. Otolaryngol. Head Neck Surg. 2017, 157 (Suppl. 2), S1–S30. [Google Scholar] [CrossRef]
- Barros, M.R., Jr.; de Melo, C.M.L.; Barros, M.; de Cassia Pereira de Lima, R.; de Freitas, A.C.; Venuti, A. Activities of stromal and immune cells in HPV-related cancers. J. Exp. Clin. Cancer Res. 2018, 37, 137. [Google Scholar] [CrossRef]
- Yang, W.; Song, Y.; Lu, Y.L.; Sun, J.Z.; Wang, H.W. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology 2013, 139, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Bashaw, A.A.; Leggatt, G.R.; Chandra, J.; Tuong, Z.K.; Frazer, I.H. Modulation of antigen presenting cell functions during chronic HPV infection. Papillomavirus Res. 2017, 4, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Pahne-Zeppenfeld, J.; Schroer, N.; Walch-Ruckheim, B.; Oldak, M.; Gorter, A.; Hegde, S.; Smola, S. Cervical cancer cell-derived interleukin-6 impairs CCR7-dependent migration of MMP-9-expressing dendritic cells. Int. J. Cancer 2014, 134, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Khallouf, H.; Grabowska, A.K.; Riemer, A.B. Therapeutic Vaccine Strategies against Human Papillomavirus. Vaccines 2014, 2, 422–462. [Google Scholar] [CrossRef]
- Le Poole, I.C.; ElMasri, W.M.; Denman, C.J.; Kroll, T.M.; Bommiasamy, H.; Lyons Eiben, G.; Kast, W.M. Langerhans cells and dendritic cells are cytotoxic towards HPV16 E6 and E7 expressing target cells. Cancer Immunol. Immunother. 2008, 57, 789–797. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, K.; Huang, Y.; Zhang, H.; Zhou, L.; Li, A.; Sun, Y. Photosensitizer-induced HPV16 E7 nanovaccines for cervical cancer immunotherapy. Biomaterials 2022, 282, 121411. [Google Scholar] [CrossRef]



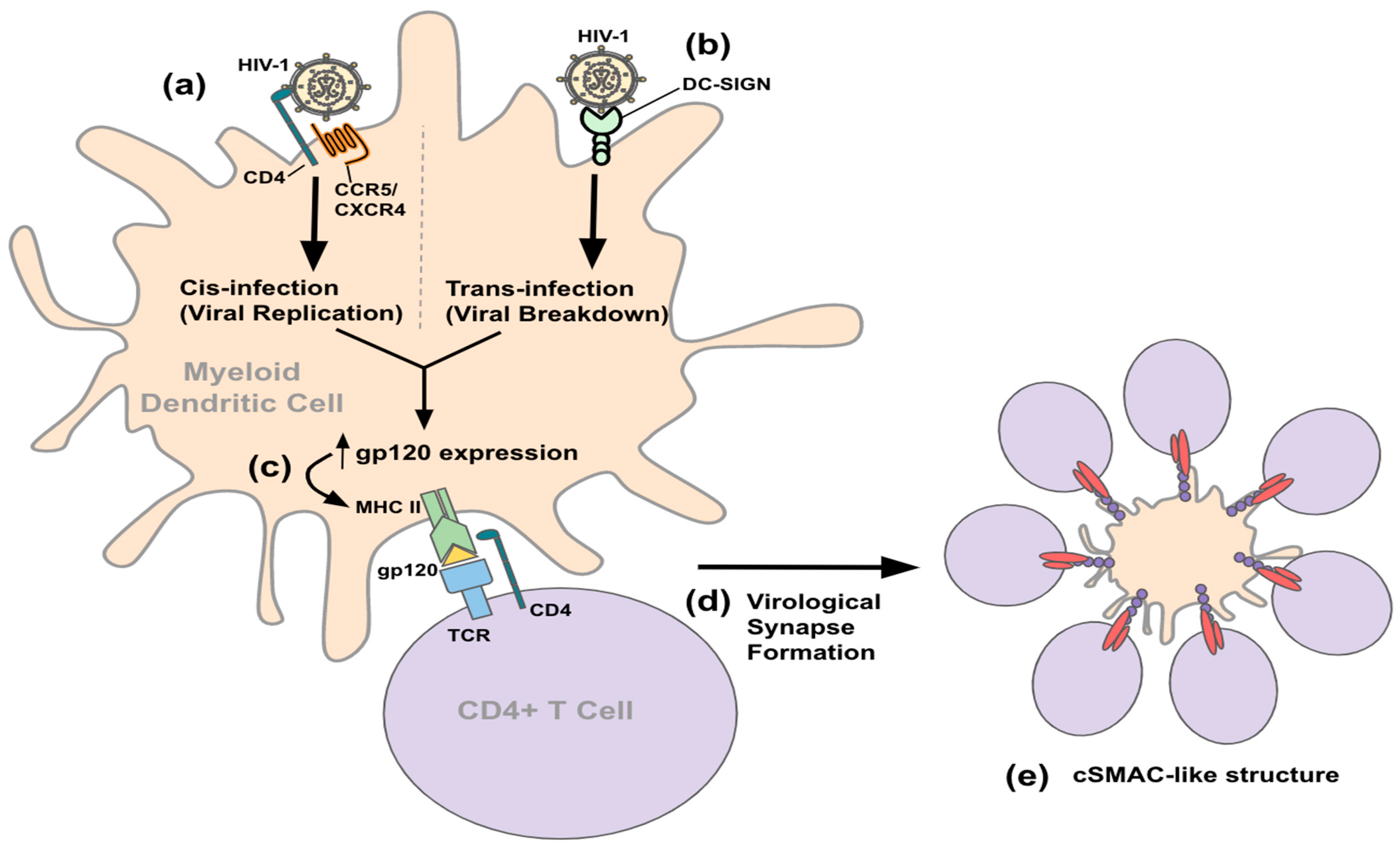{kind=link}
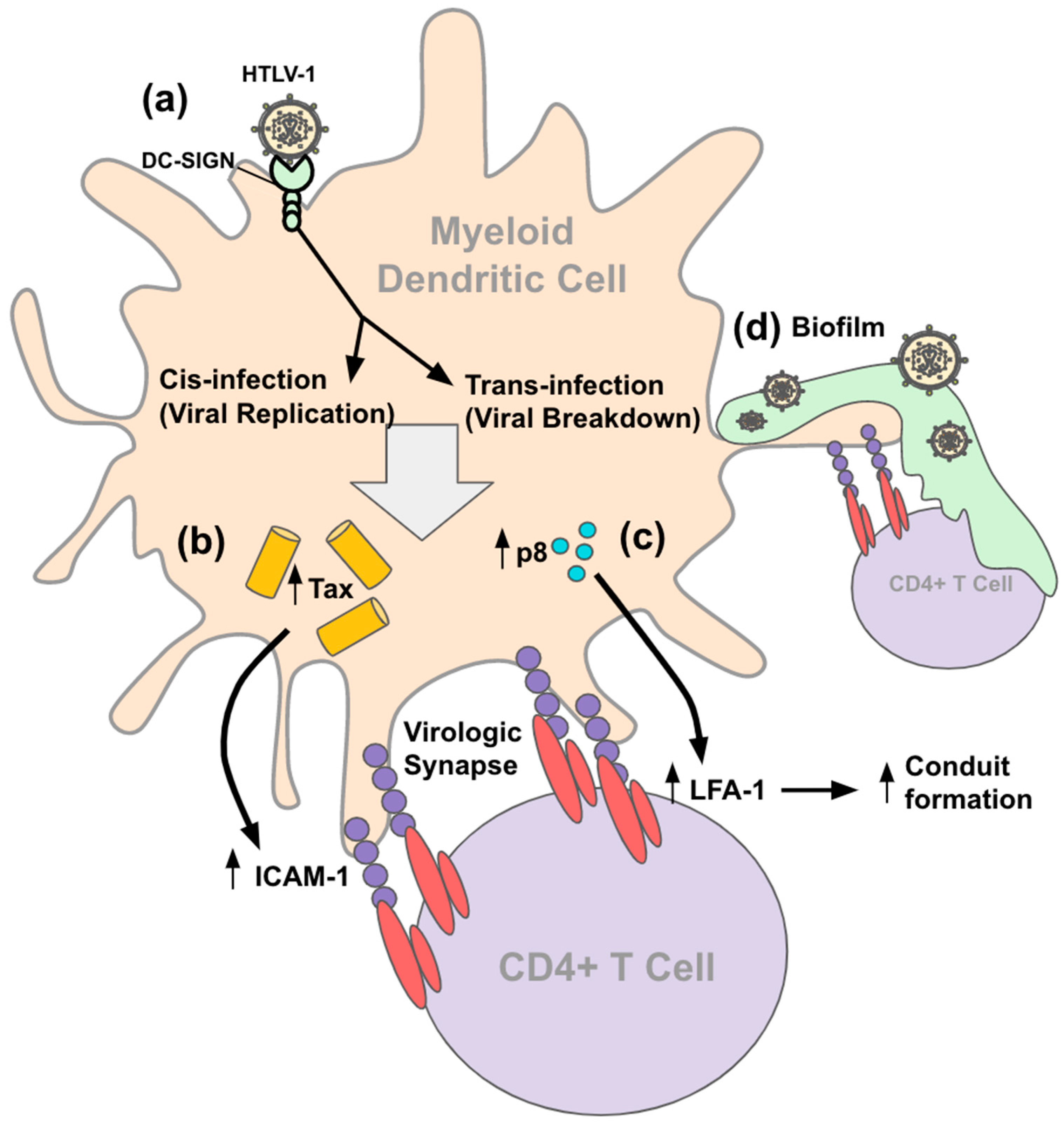{kind=link}
| DC Location | CD Molecule Expression | Cytokines and Signaling Molecules |
|---|---|---|
| Lymph Nodes | CD80, CD86, CD40 | CCR7, CCL19/CCL21, CXCL9/10, |
| Spleen | CD8, Esam(hi)CD11b+, CD205, CD207, Clec9a | Proliferate in presence of Flt3L, Notch2 |
| Thymus | CD8, CD8a, CD207, CD1a, CD11c | SIRPα+ dependent on CCR2 chemotaxis, S100, |
| Blood (pDC, cDC, iDC) | pDC: BDCA2, BDCA4 cDC: CD11 | CCR7, CCR2, CXCR3, MHCI, IL-12, IL-2, IL-15, CCL3, CXCL8, TNF |
| Skin (Langerhans) | CD207, CD45, CD11c, CD1a, CD14, CD141 | Proliferate independent of Flt3; Dependent on CSF-1R which induces chemokines CCL2 & CCL20, CCR7, CCL21, CCR2, IFN-λ, TLR3, IFN-α/β, IL-12p40, XCR1, IDO, TLR2/8, CD5, TLR7 |
| Gut | CD8, CD103, CD207, CD11b, CD70 | Proliferate in presence of Flt3L, Notch2, ALDH1A1/2, RA, IL-5, IL-6, TGFβ, CX3CR1, TLR5, Flagellin, TSLP, IL-23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulherkar, T.H.; Gómez, D.J.; Sandel, G.; Jain, P. Co-Infection and Cancer: Host–Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses. Viruses 2022, 14, 2037. https://doi.org/10.3390/v14092037
Mulherkar TH, Gómez DJ, Sandel G, Jain P. Co-Infection and Cancer: Host–Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses. Viruses. 2022; 14(9):2037. https://doi.org/10.3390/v14092037
Chicago/Turabian StyleMulherkar, Tania H., Daniel Joseph Gómez, Grace Sandel, and Pooja Jain. 2022. "Co-Infection and Cancer: Host–Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses" Viruses 14, no. 9: 2037. https://doi.org/10.3390/v14092037
APA StyleMulherkar, T. H., Gómez, D. J., Sandel, G., & Jain, P. (2022). Co-Infection and Cancer: Host–Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses. Viruses, 14(9), 2037. https://doi.org/10.3390/v14092037