
Next Generation Sequencing for the Analysis of Parvovirus B19 Genomic Diversity

 ,
,  ,
,
Abstract
:1. Introduction
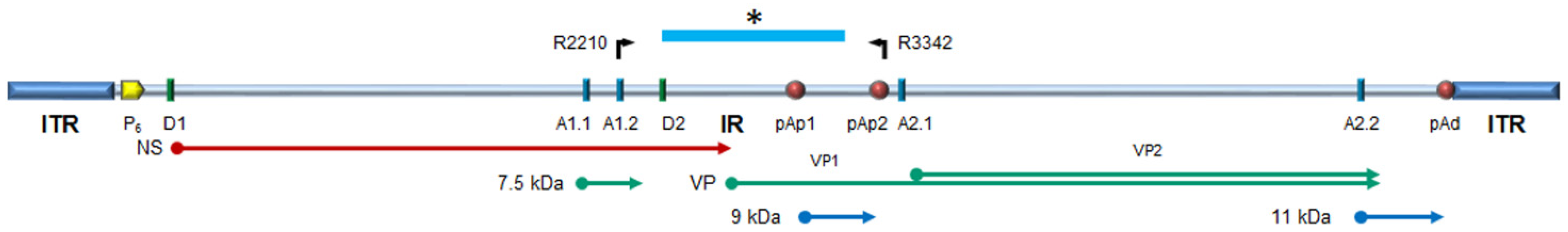
2. Materials and Methods
3. Results
3.1. Samples and NGS Output
3.2. Sequence Alignment
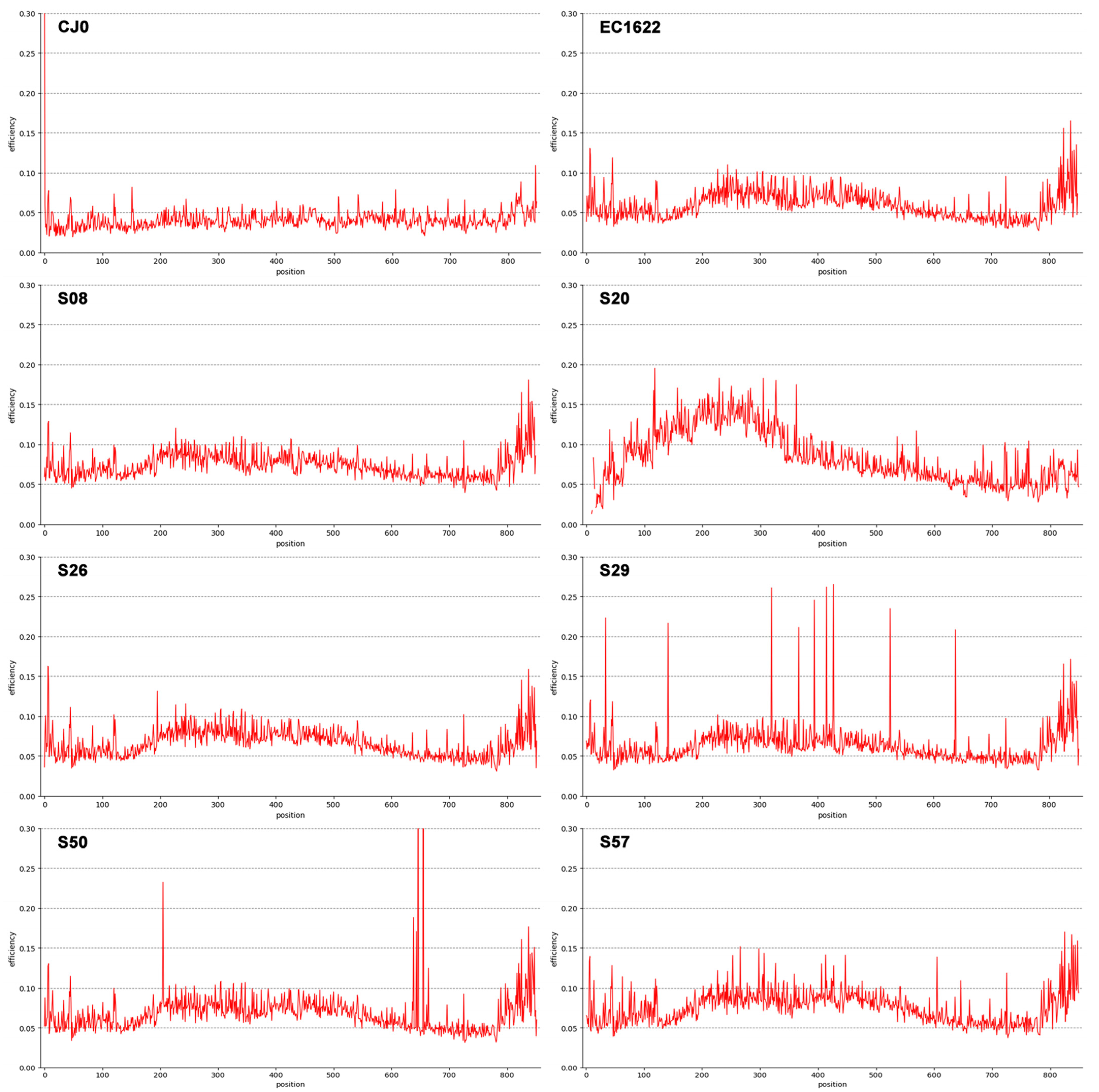
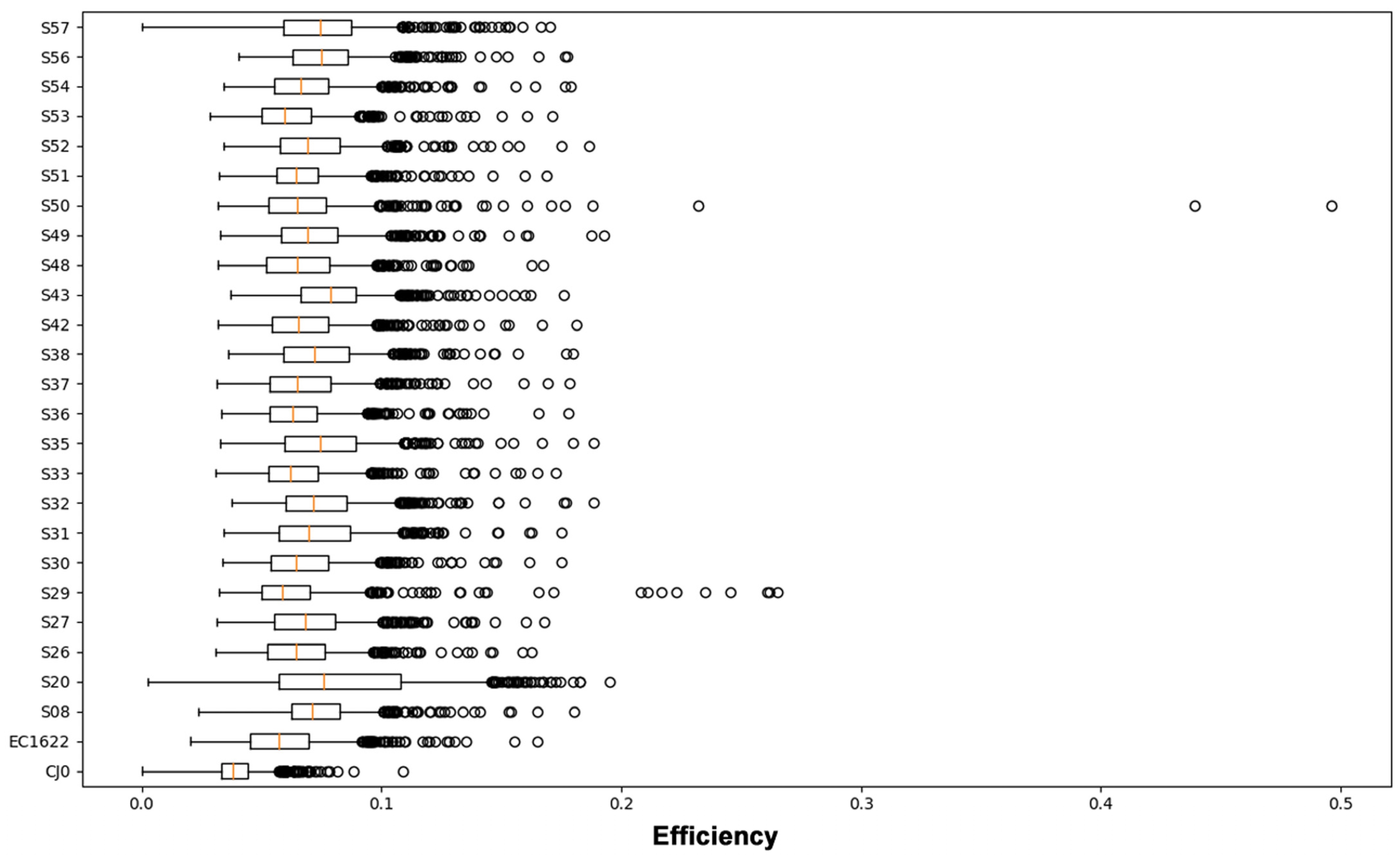
3.3. Sequence Variability Analysis
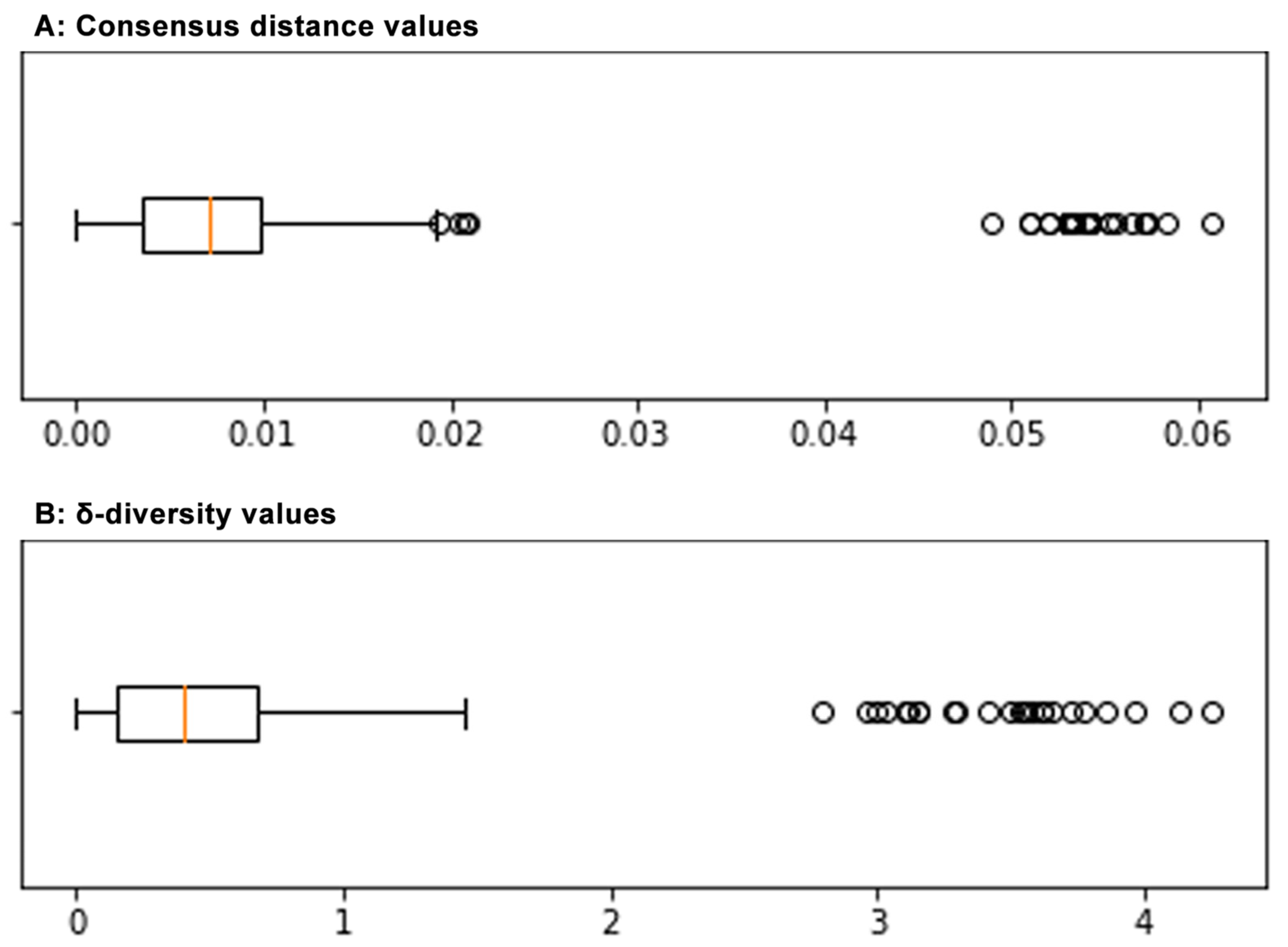
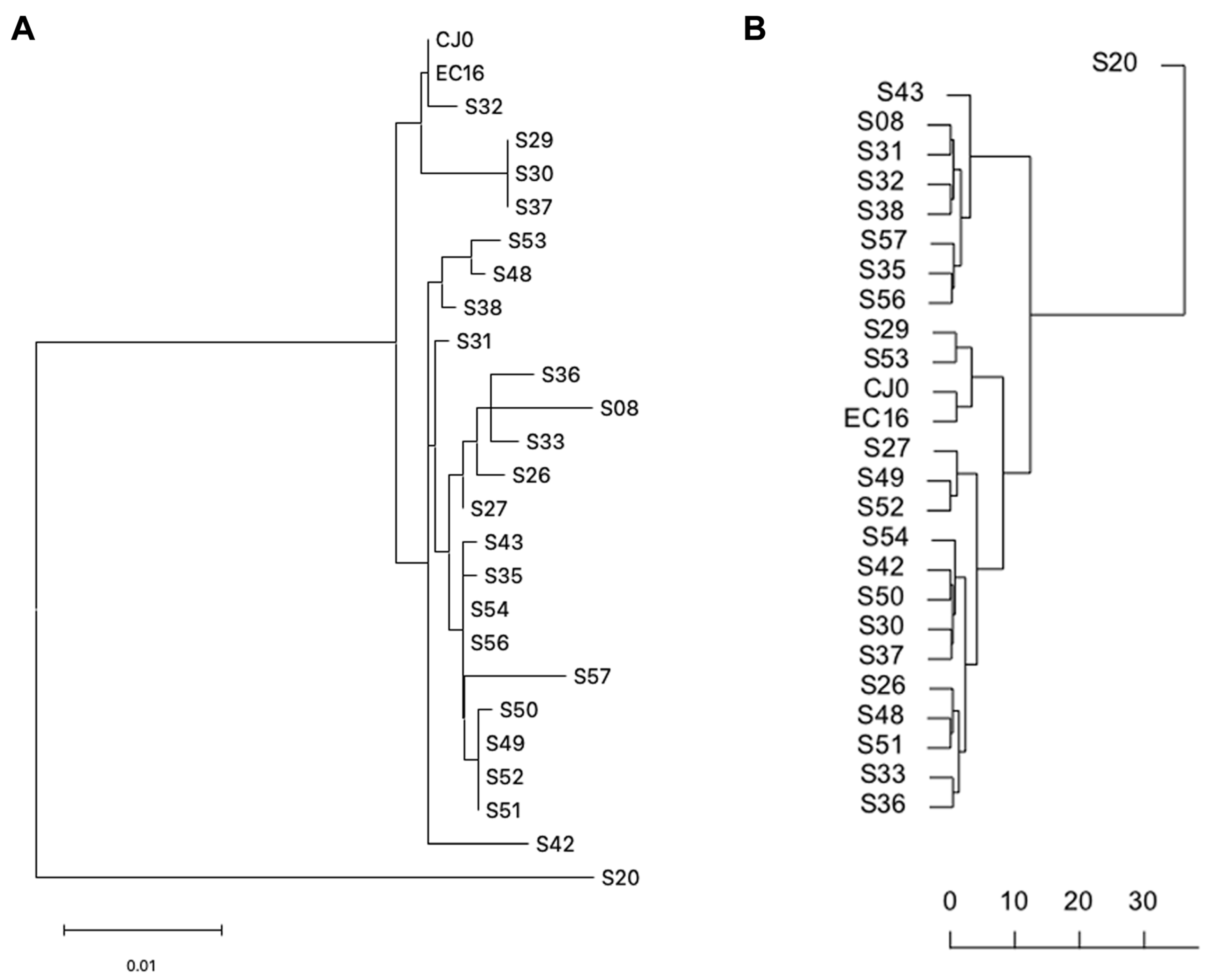
3.4. Sequence Distances Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallinella, G. Parvoviridae. In Encyclopedia of Infection and Immunity; Rezaei, N., Ed.; Elsevier: Oxford, UK, 2022; pp. 259–277. [Google Scholar] [CrossRef]
- Bua, G.; Manaresi, E.; Bonvicini, F.; Gallinella, G. Parvovirus B19 Replication and Expression in Differentiating Erythroid Progenitor Cells. PLoS ONE 2016, 11, e0148547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.E. Haematological consequences of parvovirus B19 infection. Baillieres Clin. Haematol. 2000, 13, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R. A review of blood diseases and cytopenias associated with human parvovirus B19 infection. Rev. Med. Virol. 2015, 25, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Bua, G.; Gallinella, G. How does parvovirus B19 DNA achieve lifelong persistence in human cells? Futur. Virol. 2017, 12, 549–553. [Google Scholar] [CrossRef]
- Adamson-Small, L.A.; Ignatovich, I.V.; Laemmerhirt, M.G.; Hobbs, J.A. Persistent parvovirus B19 infection in non-erythroid tissues: Possible role in the inflammatory and disease process. Virus Res. 2014, 190, 8–16. [Google Scholar] [CrossRef]
- Young, N.S.; Brown, K.E. Parvovirus B19. N. Engl. J. Med. 2004, 350, 586–597. [Google Scholar] [CrossRef]
- Servant, A.; Laperche, S.; Lallemand, F.; Marinho, V.; De Saint Maur, G.; Meritet, J.F.; Garbarg-Chenon, A. Genetic diversity within human erythroviruses: Identification of three genotypes. J. Virol. 2002, 76, 9124–9134. [Google Scholar] [CrossRef] [Green Version]
- Gallinella, G.; Venturoli, S.; Manaresi, E.; Musiani, M.; Zerbini, M. B19 virus genome diversity: Epidemiological and clinical correlations. J. Clin. Virol. 2003, 28, 1–13. [Google Scholar] [CrossRef]
- Hübschen, J.M.; Mihneva, Z.; Mentis, A.F.; Schneider, F.; Aboudy, Y.; Grossman, Z.; Rudich, H.; Kasymbekova, K.; Sarv, I.; Nedeljkovic, J.; et al. Phylogenetic analysis of human parvovirus B19 sequences from eleven different countries confirms the predominance of genotype 1 and suggests the spread of genotype 3b. J. Clin. Microbiol. 2009, 47, 3735–3738. [Google Scholar] [CrossRef]
- Norja, P.; Hokynar, K.; Aaltonen, L.-M.; Chen, R.; Ranki, A.; Partio, E.K.; Kiviluoto, O.; Davidkin, I.; Leivo, T.; Eis-Hübinger, A.M.; et al. Bioportfolio: Lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 7450–7453. [Google Scholar] [CrossRef] [Green Version]
- Eis-Hübinger, A.M.; Reber, U.; Edelmann, A.; Kalus, U.; Hofmann, J. Parvovirus B19 genotype 2 in blood donations. Transfusion 2014, 54, 1682–1684. [Google Scholar] [CrossRef]
- Bonvicini, F.; Manaresi, E.; Bua, G.; Venturoli, S.; Gallinella, G. Keeping pace with parvovirus B19 genetic variability: A multiplex genotype-specific quantitative PCR assay. J. Clin. Microbiol. 2013, 51, 3753–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Guan, W.; Cheng, F.; Chen, A.Y.; Qiu, J. Molecular characterization of human parvovirus B19 genotypes 2 and 3. Virology 2009, 394, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekman, A.; Hokynar, K.; Kakkola, L.; Kantola, K.; Hedman, L.; Bondén, H.; Gessner, M.; Aberham, C.; Norja, P.; Miettinen, S.; et al. Biological and immunological relations among human parvovirus B19 genotypes 1 to 3. J. Virol. 2007, 81, 6927–6935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyöriä, L.; Toppinen, M.; Mäntylä, E.; Hedman, L.; Aaltonen, L.M.; Vihinen-Ranta, M.; Ilmarinen, T.; Söderlund-Venermo, M.; Hedman, K.; Perdomo, M.F. Extinct type of human parvovirus B19 persists in tonsillar B cells. Nat. Commun. 2017, 8, 14930. [Google Scholar] [CrossRef] [Green Version]
- Norja, P.; Eis-Hübinger, A.M.; Söderlund-Venermo, M.; Hedman, K.; Simmonds, P. Rapid sequence change and geographical spread of human parvovirus B19: Comparison of B19 virus evolution in acute and persistent infections. J. Virol. 2008, 82, 6427–6433. [Google Scholar] [CrossRef] [Green Version]
- Stamenković, G.G.; Cirkovic, V.; Siljic, M.; Blagojevic, J.; Knezevic, A.; Joksić, I.D.; Stanojević, M.P. Substitution rate and natural selection in parvovirus B19. Sci. Rep. 2016, 6, 35759. [Google Scholar] [CrossRef] [Green Version]
- Mühlemann, B.; Margaryan, A.; Damgaard, P.D.B.; Allentoft, M.E.; Vinner, L.; Hansen, A.J.; Weber, A.; Bazaliiskii, V.I.; Molak, M.; Arneborg, J.; et al. Ancient human parvovirus B19 in Eurasia reveals its long-term association with humans. Proc. Natl. Acad. Sci. USA 2018, 115, 7557–7562. [Google Scholar] [CrossRef] [Green Version]
- Guzmán-Solís, A.A.; Villa-Islas, V.; Bravo-López, M.J.; Sandoval-Velasco, M.; Wesp, J.K.; Gómez-Valdés, J.A.; Moreno-Cabrera, M.D.L.L.; Meraz, A.; Solís-Pichardo, G.; Schaaf, P.; et al. Ancient viral genomes reveal introduction of human pathogenic viruses into Mexico during the transatlantic slave trade. elife 2021, 10. [Google Scholar] [CrossRef]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Beerenwinkel, N.; Zagordi, O. Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol. 2011, 1, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2016, 239, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manaresi, E.; Conti, I.; Bua, G.; Bonvicini, F.; Gallinella, G. A Parvovirus B19 synthetic genome: Sequence features and functional competence. Virology 2017, 508, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, F.; Filippone, C.; Delbarba, S.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Parvovirus B19 genome as a single, two-state replicative and transcriptional unit. Virology 2006, 347, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, F.; Filippone, C.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Functional analysis and quantitative determination of the expression profile of human parvovirus B19. Virology 2008, 381, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Manaresi, E.; Gallinella, G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses 2019, 11, 659. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-throughput sequencing (HTS) for the analysis of viral populations. Infect. Genet. Evol. 2020, 80, 104208. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Freimanis, G.; Lasecka-Dykes, L.; Asfor, A.; Ribeca, P.; Waters, R.; King, D.; Laing, E. A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations. Viruses 2020, 12, 1187. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, M.C.F.; Yin, L.; Nolan, D.J.; Lowe, A.D.; Goodenow, M.M.; Salemi, M. Empirical validation of viral quasispecies assembly algorithms: State-of-the-art and challenges. Sci. Rep. 2013, 3, 2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, I.N.; Muller, C.P.; He, F.Q. Applying next-generation sequencing to unravel the mutational landscape in viral quasispecies. Virus Res. 2020, 283, 197963. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Perales, C.; Rodriguez-Frias, F.; Esteban, J.I.; Quer, J.; Domingo, E. Viral quasispecies complexity measures. Virology 2016, 493, 227–237. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of war—Host adaptation and its constraints on virus evolution. Nat. Rev. Microbiol. 2019, 17, 321–328. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Date | Viral Load | Variations 2 | Distance to CJ0 3 | Mean Distance 3,5 | α-Diversity 4 | δ-Diversity 4,5 |
|---|---|---|---|---|---|---|---|
| CJ0 | --- | 1.00 × 106 | 0 | 0.0000 | 0.0067 | 0.0403 | 0.9376 |
| EC1622 | --- | 1.00 × 106 | 0 | 0.0000 | 0.0067 | 0.0595 | 1.0601 |
| S08 | 04/09/2019 | 8.26 × 106 | 14 | 0.0127 | 0.0105 | 0.0735 | 0.4695 |
| S26 | 21/03/2012 | 1.00 × 1010 | 8 | 0.0072 | 0.0063 | 0.0663 | 0.4555 |
| S27 | 14/04/2012 | 1.20 × 108 | 6 | 0.0054 | 0.0047 | 0.0699 | 0.3891 |
| S29 | 11/06/2012 | 5.40 × 106 | 6 | 0.0053 | 0.0099 | 0.0635 | 0.6682 |
| S30 | 14/06/2012 | 5.00 × 109 | 6 | 0.0053 | 0.0099 | 0.0674 | 0.4018 |
| S31 | 15/06/2012 | 6.00 × 109 | 5 | 0.0045 | 0.0052 | 0.0737 | 0.4735 |
| S32 | 27/06/2012 | 2.00 × 106 | 2 | 0.0018 | 0.0084 | 0.0744 | 0.5090 |
| S33 | 06/11/2012 | 2.00 × 1010 | 10 | 0.0063 | 0.0050 | 0.0647 | 0.5663 |
| S35 | 06/06/2013 | 1.00 × 1010 | 7 | 0.0081 | 0.0070 | 0.0759 | 0.6108 |
| S36 | 25/06/2013 | 1.00 × 1010 | 11 | 0.0090 | 0.0079 | 0.0654 | 0.5164 |
| S37 | 12/09/2013 | 1.00 × 109 | 6 | 0.0053 | 0.0099 | 0.0677 | 0.3932 |
| S38 | 26/09/2013 | 1.00 × 107 | 4 | 0.0036 | 0.0056 | 0.0742 | 0.5011 |
| S42 | 07/05/2015 | 3.00 × 106 | 11 | 0.0099 | 0.0098 | 0.0681 | 0.3863 |
| S43 | 27/06/2015 | 1.00 × 106 | 7 | 0.0062 | 0.0050 | 0.0787 | 0.8703 |
| S48 | 27/01/2016 | 1.50 × 107 | 8 | 0.0072 | 0.0074 | 0.0670 | 0.4206 |
| S49 | 03/03/2016 | 2.00 × 106 | 7 | 0.0062 | 0.0046 | 0.0716 | 0.4116 |
| S50 | 23/08/2016 | 3.00 × 106 | 7 | 0.0072 | 0.0054 | 0.0680 | 0.3872 |
| S51 | 23/03/2017 | 1.00 × 1010 | 7 | 0.0062 | 0.0046 | 0.0669 | 0.4247 |
| S52 | 01/06/2017 | 2.00 × 108 | 7 | 0.0062 | 0.0046 | 0.0717 | 0.4125 |
| S53 | 29/07/2017 | 5.00 × 107 | 8 | 0.0081 | 0.0081 | 0.0620 | 0.8029 |
| S54 | 28/11/2018 | 1.86 × 106 | 6 | 0.0054 | 0.0042 | 0.0687 | 0.3837 |
| S56 | 26/06/2019 | 2.69 × 106 | 6 | 0.0054 | 0.0042 | 0.0763 | 0.6484 |
| S57 | 23/07/2019 | 2.96 × 106 | 13 | 0.0117 | 0.0100 | 0.0754 | 0.5745 |
| Mean for Genotype 1 | 7 | 0.0067 | 0.0069 | 0.0700 | 0.5077 | ||
| S20 | 02/05/2020 | 7.65 × 106 | 53 | 0.0509 | 0.0547 | 0.0828 | 3.0858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bichicchi, F.; Guglietta, N.; Rocha Alves, A.D.; Fasano, E.; Manaresi, E.; Bua, G.; Gallinella, G. Next Generation Sequencing for the Analysis of Parvovirus B19 Genomic Diversity. Viruses 2023, 15, 217. https://doi.org/10.3390/v15010217
Bichicchi F, Guglietta N, Rocha Alves AD, Fasano E, Manaresi E, Bua G, Gallinella G. Next Generation Sequencing for the Analysis of Parvovirus B19 Genomic Diversity. Viruses. 2023; 15(1):217. https://doi.org/10.3390/v15010217
Chicago/Turabian StyleBichicchi, Federica, Niccolò Guglietta, Arthur Daniel Rocha Alves, Erika Fasano, Elisabetta Manaresi, Gloria Bua, and Giorgio Gallinella. 2023. "Next Generation Sequencing for the Analysis of Parvovirus B19 Genomic Diversity" Viruses 15, no. 1: 217. https://doi.org/10.3390/v15010217
APA StyleBichicchi, F., Guglietta, N., Rocha Alves, A. D., Fasano, E., Manaresi, E., Bua, G., & Gallinella, G. (2023). Next Generation Sequencing for the Analysis of Parvovirus B19 Genomic Diversity. Viruses, 15(1), 217. https://doi.org/10.3390/v15010217