
Evaluation of Human Mesenchymal Stromal Cells as Carriers for the Delivery of Oncolytic HAdV-5 to Head and Neck Squamous Cell Carcinomas

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Human MSC Isolation, Characterization, and Cultivation
2.3. Adenoviral Vectors
2.4. Transduction/Infection of hMSCs with HAdV-5 Vectors
2.5. HNSCC Xenograft Mouse Model
2.6. Analysis of In Vivo hMSC Biodistribution Using the IVIS 200 System
2.7. Tissue Preparation for Immunohistochemistry
2.8. Analysis of hMSC Migration in the Chorioallantoic Membrane (CAM) Model
2.9. Immunohistochemical Staining of CD31
2.10. DNA Extraction from Tissue Samples and Analysis of Adenoviral E4 Copy Number Using Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.11. Virus Replication Assay
2.12. Quantification of Virus Replication in hMSCs by qPCR
2.13. Analysis of Lactate Dehydrogenase (LDH) Release to Cell Culture Supernatant
2.14. Analysis of Virus Spread by Plaque Assays
2.15. Migration Assays In Vitro
2.16. Statistical Analysis
3. Results
3.1. Replication-Competent HAdV-5-HexPos3 Particles Efficiently Replicate in BM- and A-hMSCs
3.2. Replication-Competent HAdV-5-HexPos3 Vectors Carrying the E1B-Δ19K Mutation Show Accelerated Replication and Release of Mature Viral Progeny in hMSCs
3.3. Migration of hMSCs Is Not Inhibited by Virus Infection In Vitro
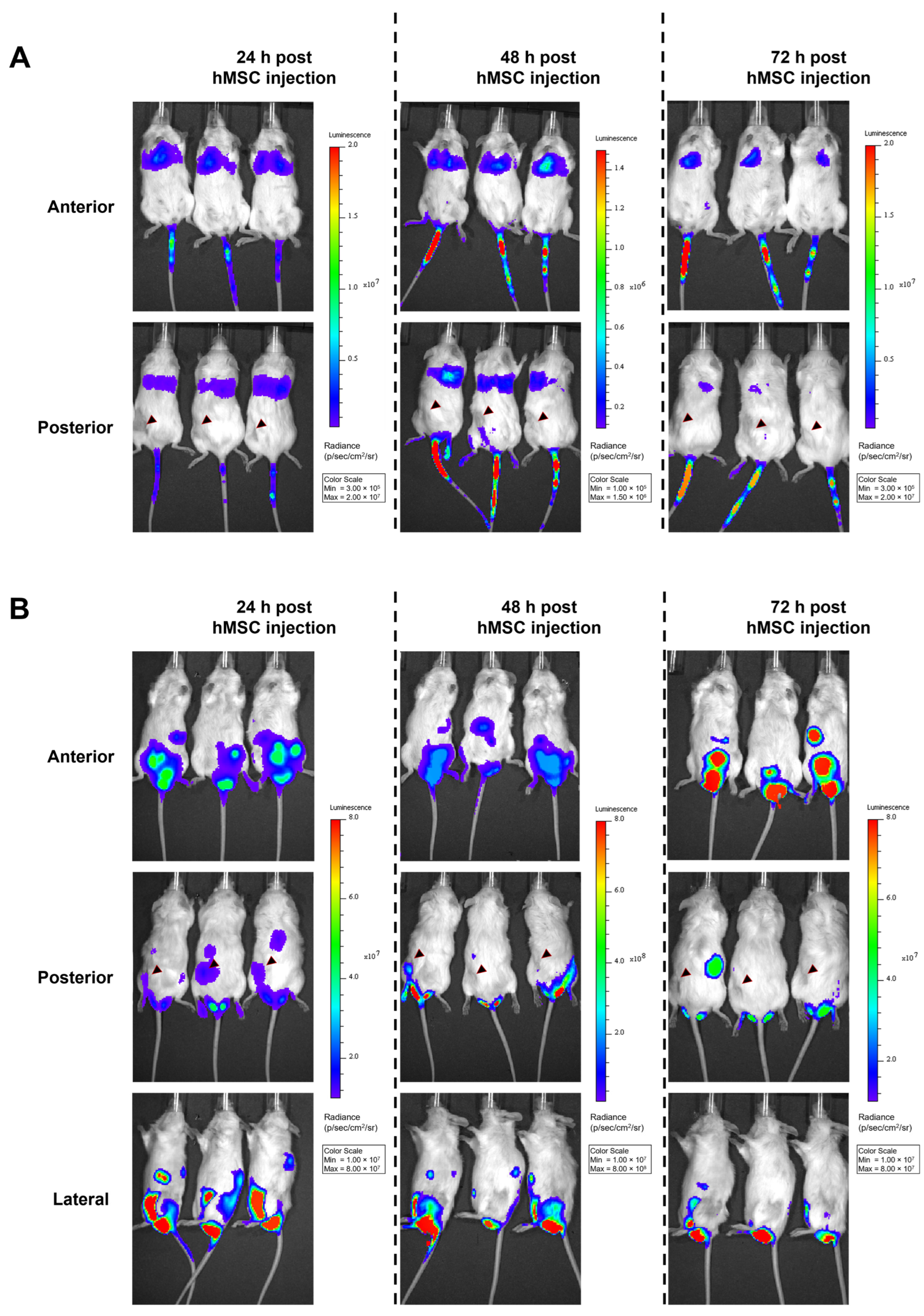
3.4. No Migration of hMSCs toward a Xenograft HNSCC Tumor In Vivo
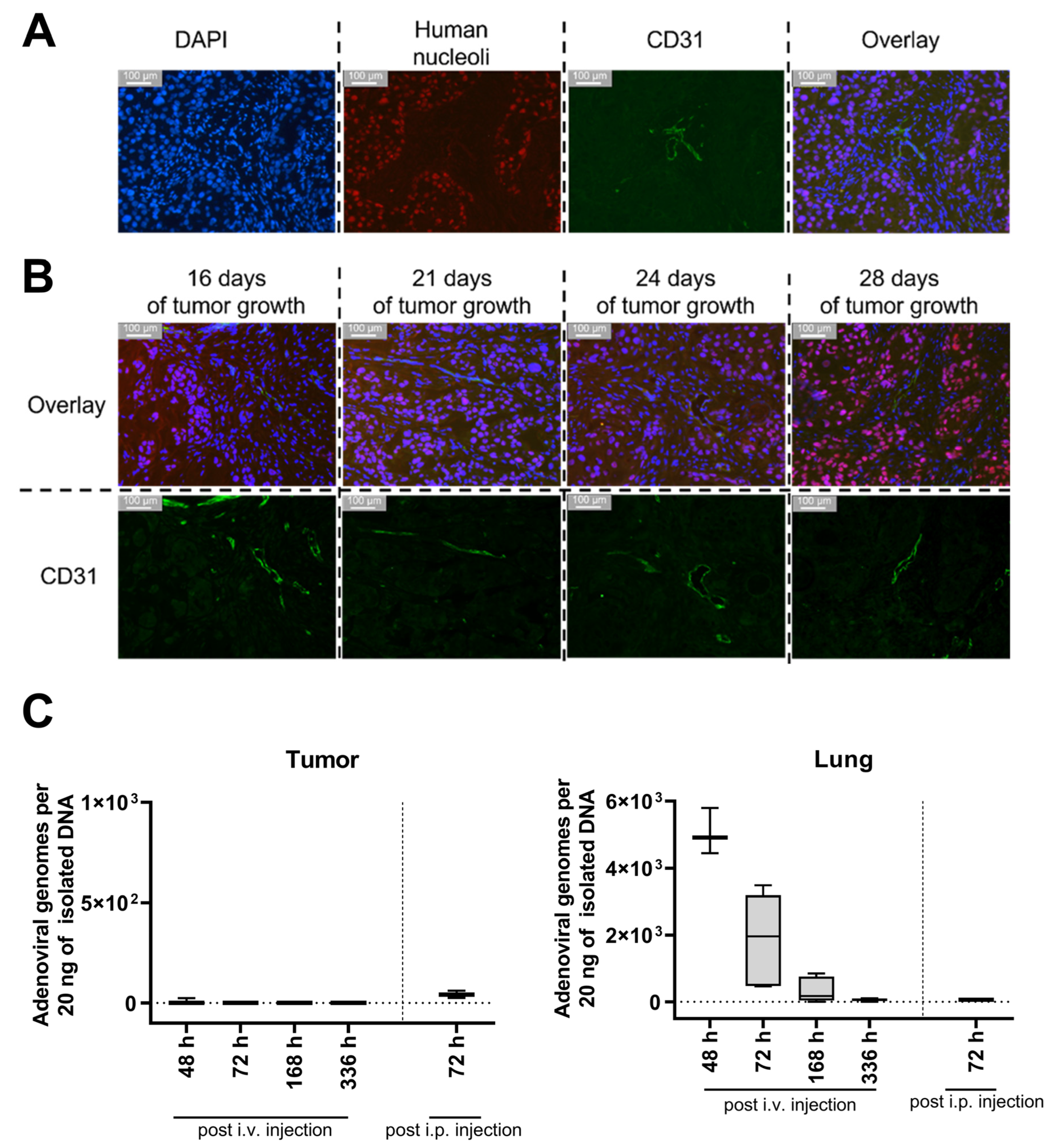
3.5. No Migration of hMSCs toward a Xenograft HNSCC Tumor in a Chorioallantoic Membrane Model
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Head and Neck Cancer: Statistics. Available online: https://www.cancer.net (accessed on 26 October 2022).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, M.; Yura, Y. Efficient Delivery and Replication of Oncolytic Virus for Successful Treatment of Head and Neck Cancer. Int. J. Mol. Sci. 2020, 21, 7073. [Google Scholar] [CrossRef]
- Dias, J.D.; Guse, K.; Nokisalmi, P.; Eriksson, M.; Chen, D.-T.; Diaconu, I.; Tenhunen, M.; Liikanen, I.; Grénman, R.; Savontaus, M.; et al. Multimodal Approach Using Oncolytic Adenovirus, Cetuximab, Chemotherapy and Radiotherapy in HNSCC Low Passage Tumour Cell Cultures. Eur. J. Cancer 2010, 46, 625–635. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Han, J.; Salzwedel, A.O.; Davydova, J.; Herzberg, M.C.; Gopalakrishnan, R.; Yamamoto, M. Oncolytic Adenoviruses Targeted to Human Papilloma Virus-Positive Head and Neck Squamous Cell Carcinomas. Oral Oncol. 2016, 56, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Vijayalingam, S.; Kuppuswamy, M.; Subramanian, T.; Strebeck, F.F.; West, C.L.; Varvares, M.; Chinnadurai, G. Evaluation of Apoptogenic Adenovirus Type 5 Oncolytic Vectors in a Syrian Hamster Head and Neck Cancer Model. Cancer Gene Ther. 2014, 21, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Seiradake, E.; Henaff, D.; Wodrich, H.; Billet, O.; Perreau, M.; Hippert, C.; Mennechet, F.; Schoehn, G.; Lortat-Jacob, H.; Dreja, H.; et al. The Cell Adhesion Molecule “CAR” and Sialic Acid on Human Erythrocytes Influence Adenovirus in Vivo Biodistribution. PLoS Pathog. 2009, 5, e1000277. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.P.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.C.; Fisher, K.D.; et al. Human Erythrocytes Bind and Inactivate Type 5 Adenovirus by Presenting Coxsackie Virus-Adenovirus Receptor and Complement Receptor 1. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation Factor X Shields Adenovirus Type 5 from Attack by Natural Antibodies and Complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef]
- Qiu, Q.; Xu, Z.; Tian, J.; Moitra, R.; Gunti, S.; Notkins, A.L.; Byrnes, A.P. Impact of Natural IgM Concentration on Gene Therapy with Adenovirus Type 5 Vectors. J. Virol. 2014, 89, 3412–3416. [Google Scholar] [CrossRef] [PubMed]
- Cichon, G.; Boeckh-Herwig, S.; Schmidt, H.H.; Wehnes, E.; Müller, T.; Pring-Akerblom, P.; Burger, R. Complement Activation by Recombinant Adenoviruses. Gene Ther. 2001, 8, 1794–1800. [Google Scholar] [CrossRef] [Green Version]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International Epidemiology of Human Pre-Existing Adenovirus (Ad) Type-5, Type-6, Type-26 and Type-36 Neutralizing Antibodies: Correlates of High Ad5 Titers and Implications for Potential HIV Vaccine Trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef]
- Waddington, S.N.; Parker, A.L.; Havenga, M.; Nicklin, S.A.; Buckley, S.M.K.; McVey, J.H.; Baker, A.H. Targeting of Adenovirus Serotype 5 (Ad5) and 5/47 Pseudotyped Vectors In Vivo: Fundamental Involvement of Coagulation Factors and Redundancy of CAR Binding by Ad5. J. Virol. 2007, 81, 9568–9571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.-Y.; Lieber, A. Adenovirus Binding to Blood Factors Results in Liver Cell Infection and Hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, R.; Reddy, V.S.; Nemerow, G.R.; Barry, M.A. Identification of Adenovirus Serotype 5 Hexon Regions That Interact with Scavenger Receptors. J. Virol. 2012, 86, 2293–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer Cell-Evading Adenovirus for Systemic and Liver-Directed Gene Transfer. Mol. Ther. 2011, 19, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Haisma, H.J.; Boesjes, M.; Beerens, A.M.; Van Der Strate, B.W.A.; Curiel, D.T.; Plüddemann, A.; Gordon, S.; Bellu, A.R. Scavenger Receptor A: A New Route for Adenovirus 5. Mol. Pharm. 2009, 6, 366–374. [Google Scholar] [CrossRef]
- Goradel, N.H.; Negahdari, B.; Ghorghanlu, S.; Jahangiri, S.; Arashkia, A. Strategies for Enhancing Intratumoral Spread of Oncolytic Adenoviruses. Pharmacol. Ther. 2020, 213, 107586. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [Green Version]
- Moreno, R. Mesenchymal Stem Cells and Oncolytic Viruses: Joining Forces against Cancer. J. Immunother. Cancer 2021, 9, e001684. [Google Scholar] [CrossRef]
- Ruano, D.; López-Martín, J.A.; Moreno, L.; Lassaletta, Á.; Bautista, F.; Andión, M.; Hernández, C.; González-Murillo, Á.; Melen, G.; Alemany, R.; et al. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol. Ther. 2020, 28, 1033–1042. [Google Scholar] [CrossRef]
- Nilson, R.; Lübbers, O.; Weiß, L.; Singh, K.; Scharffetter-Kochanek, K.; Rojewski, M.; Schrezenmeier, H.; Zeplin, P.H.; Funk, W.; Krutzke, L.; et al. Transduction Enhancers Enable Efficient Human Adenovirus Type 5-Mediated Gene Transfer into Human Multipotent Mesenchymal Stromal Cells. Viruses 2021, 13, 1136. [Google Scholar] [CrossRef]
- Reynolds, P.N.; Dmitriev, I.; Curiel, D.T. Insertion of an RGD Motif into the HI Loop of Adenovirus Fiber Protein Alters the Distribution of Transgene Expression of the Systemically Administered Vector. Gene Ther. 1999, 6, 1336–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, R.; Rojas, L.A.; Villellas, F.V.; Soriano, V.C.; García-Castro, J.; Fajardo, C.A.; Alemany, R. Human Menstrual Blood-Derived Mesenchymal Stem Cells as Potential Cell Carriers for Oncolytic Adenovirus. Stem Cells Int. 2017, 2017, 3615729. [Google Scholar] [CrossRef] [Green Version]
- Hammer, K.; Kazcorowski, A.; Liu, L.; Behr, M.; Schemmer, P.; Herr, I.; Nettelbeck, D.M. Engineered Adenoviruses Combine Enhanced Oncolysis with Improved Virus Production by Mesenchymal Stromal Carrier Cells. Int. J. Cancer 2015, 137, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 Is a Receptor for Adenovirus Serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Sirena, D.; Lilienfeld, B.; Eisenhut, M.; Kälin, S.; Boucke, K.; Beerli, R.R.; Vogt, L.; Ruedl, C.; Bachmann, M.F.; Greber, U.F.; et al. The Human Membrane Cofactor CD46 Is a Receptor for Species B Adenovirus Serotype 3. J. Virol. 2004, 78, 4454–4462. [Google Scholar] [CrossRef] [Green Version]
- Nilson, R.; Lübbers, O.; Schmidt, C.Q.; Rojewski, M.; Zeplin, P.H.; Funk, W.; Schrezenmeier, H.; Kritzinger, A.; Kochanek, S.; Krutzke, L. Hexon Modification of Human Adenovirus Type 5 Vectors Enables Efficient Transduction of Human Multipotent Mesenchymal Stromal Cells. Mol. Ther.-Methods Clin. Dev. 2022, 25, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Hertel, S.; Kochanek, S. Efficient Transformation of Primary Human Amniocytes by E1 Functions of Ad5: Generation of New Cell Lines for Adenoviral Vector Production. Hum. Gene Ther. 2000, 11, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Fekete, N.; Gadelorge, M.; Frst, D.; Maurer, C.; Dausend, J.; Fleury-Cappellesso, S.; Mailnder, V.; Lotfi, R.; Ignatius, A.; Sensebé, L.; et al. Platelet Lysate from Whole Blood-Derived Pooled Platelet Concentrates and Apheresis-Derived Platelet Concentrates for the Isolation and Expansion of Human Bone Marrow Mesenchymal Stromal Cells: Production Process, Content and Identification of Active Components. Cytotherapy 2012, 14, 540–554. [Google Scholar] [CrossRef]
- Rojewski, M.T.; Lotfi, R.; Gjerde, C.; Mustafa, K.; Veronesi, E.; Ahmed, A.B.; Wiesenth, M.; Körper, S.; Sensebé, L.; Layrolle, P.; et al. Translation of a Standardized Manufacturing Protocol for Mesenchymal Stromal Cells: A Systematic Comparison of Validation and Manufacturing Data. Cytotherapy 2019, 21, 468–482. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal Cells from the Adipose Tissue-Derived Stromal Vascular Fraction and Culture Expanded Adipose Tissue-Derived Stromal/Stem Cells: A Joint Statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.-X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A Mutant Oncolytic Adenovirus Targeting the Rb Pathway Produces Anti-Glioma Effect in Vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-C.; Hallden, G.; Wang, Y.; Brooks, G.; Francis, J.; Lemoine, N.; Kirn, D. An E1B-19 KDa Gene Deletion Mutant Adenovirus Demonstrates Tumor Necrosis Factor-Enhanced Cancer Selectivity and Enhanced Oncolytic Potency. Mol. Ther. 2004, 9, 786–803. [Google Scholar] [CrossRef]
- Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.C.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of Contact Residues and Definition of the CAR-Binding Site of Adenovirus Type 5 Fiber Protein. J. Virol. 2000, 74, 2804–2813. [Google Scholar] [CrossRef] [Green Version]
- Mittereder, N.; March, K.L.; Trapnell, B.C. Evaluation of the Concentration and Bioactivity of Adenovirus Vectors for Gene Therapy. J. Virol. 1996, 70, 7498–7509. [Google Scholar] [CrossRef] [Green Version]
- Blum, H.; Beier, H.; Gross, H.J. Improved Silver Staining of Plant Proteins, RNA and DNA in Polyacrylamide Gels. Electrophoresis 1987, 8, 93–99. [Google Scholar] [CrossRef]
- Krutzke, L.; Allmendinger, E.; Hirt, K.; Kochanek, S. Chorioallantoic Membrane Tumor Model for Evaluating Oncolytic Viruses. Hum. Gene Ther. 2020, 31, 1100–1113. [Google Scholar] [CrossRef]
- Sauthoff, H.; Heitner, S.; Rom, W.N.; Hay, J.G. Deletion of the Adenoviral E1b-19kD Gene Enhances Tumor Cell Killing of a Replicating Adenoviral Vector. Available online: https://www.liebertpub.com/doi/10.1089/10430340050015851 (accessed on 6 July 2022).
- Subramanian, T.; Vijayalingam, S.; Chinnadurai, G. Genetic Identification of Adenovirus Type 5 Genes That Influence Viral Spread. J. Virol. 2006, 80, 2000–2012. [Google Scholar] [CrossRef]
- Leitner, S.; Sweeney, K.; Öberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Halldén, G. Oncolytic Adenoviral Mutants with E1B19K Gene Deletions Enhance Gemcitabine-Induced Apoptosis in Pancreatic Carcinoma Cells and Anti-Tumor Efficacy In Vivo. Clin. Cancer Res. 2009, 15, 1730–1740. [Google Scholar] [CrossRef] [Green Version]
- Rohmer, S.; Quirin, C.; Hesse, A.; Sandmann, S.; Bayer, W.; Herold-Mende, C.; Haviv, Y.S.; Wildner, O.; Enk, A.H.; Nettelbeck, D.M. Transgene Expression by Oncolytic Adenoviruses Is Modulated by E1B19K Deletion in a Cell Type-Dependent Manner. Virology 2009, 395, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.K.; Tseng, C.C.; Rao, L.; White, E. Functional Complementation of the Adenovirus E1B 19-Kilodalton Protein with Bcl-2 in the Inhibition of Apoptosis in Infected Cells. J. Virol. 1994, 68, 6553–6566. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Modha, D.; White, E. The E1B 19K Protein Associates with Lamins in Vivo and Its Proper Localization Is Required for Inhibition of Apoptosis. Oncogene 1997, 15, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Broers, J.L.V.; Bronnenberg, N.M.H.J.; Kuijpers, H.J.H.; Schutte, B.; Hutchison, C.J.; Ramaekers, F.C.S. Partial Cleavage of A-Type Lamins Concurs with Their Total Disintegration from the Nuclear Lamina during Apoptosis. Eur. J. Cell Biol. 2002, 81, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Georgi, F.; Greber, U.F. The Adenovirus Death Protein—A Small Membrane Protein Controls Cell Lysis and Disease. FEBS Lett. 2020, 594, 1861–1878. [Google Scholar] [CrossRef] [PubMed]
- Hakkarainen, T.; Särkioja, M.; Lehenkari, P.; Miettinen, S.; Ylikomi, T.; Suuronen, R.; Desmond, R.A.; Kanerva, A.; Hemminki, A. Human Mesenchymal Stem Cells Lack Tumor Tropism but Enhance the Antitumor Activity of Oncolytic Adenoviruses in Orthotopic Lung and Breast Tumors. Hum. Gene Ther. 2007, 18, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Ji, T.; Chen, P.; Li, X.; Fang, Y.; Gao, Q.; Liao, S.; You, L.; Xu, H.; Ma, Q.; et al. Mesenchymal Stem Cells as Carriers and Amplifiers in CRAd Delivery to Tumors. Mol. Cancer 2011, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Moreno, R.; Fajardo, C.A.; Farrera-Sal, M.; Perisé-Barrios, A.J.; Morales-Molina, A.; Al-Zaher, A.A.; García-Castro, J.; Alemany, R. Enhanced Antitumor Efficacy of Oncolytic Adenovirus–Loaded Menstrual Blood–Derived Mesenchymal Stem Cells in Combination with Peripheral Blood Mononuclear Cells. Mol. Cancer Ther. 2019, 18, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Zielske, S.P.; Livant, D.L.; Lawrence, T.S. Radiation Increases Invasion of Gene-Modified Mesenchymal Stem Cells into Tumors. Int. J. Radiat. Oncol. *Biol. *Phys. 2009, 75, 843–853. [Google Scholar] [CrossRef]
- Morales-Molina, Á.; Gambera, S.; Cejalvo, T.; Moreno, R.; Rodríguez-Milla, M.Á.; Perisé-Barrios, A.J.; García-Castro, J. Antitumor Virotherapy Using Syngeneic or Allogeneic Mesenchymal Stem Cell Carriers Induces Systemic Immune Response and Intratumoral Leukocyte Infiltration in Mice. Cancer Immunol. Immunother. 2018, 67, 1589–1602. [Google Scholar] [CrossRef]
- Kaczorowski, A.; Hammer, K.; Liu, L.; Villhauer, S.; Nwaeburu, C.; Fan, P.; Zhao, Z.; Gladkich, J.; Groß, W.; Nettelbeck, D.M.; et al. Delivery of Improved Oncolytic Adenoviruses by Mesenchymal Stromal Cells for Elimination of Tumorigenic Pancreatic Cancer Cells. Oncotarget 2016, 7, 9046–9059. [Google Scholar] [CrossRef] [Green Version]
- De Becker, A.; Riet, I.V. Homing and Migration of Mesenchymal Stromal Cells: How to Improve the Efficacy of Cell Therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Devine, S.M.; Cobbs, C.; Jennings, M.; Bartholomew, A.; Hoffman, R. Mesenchymal Stem Cells Distribute to a Wide Range of Tissues Following Systemic Infusion into Nonhuman Primates. Blood 2003, 101, 2999–3001. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; DiCorleto, P.E.; et al. Effect of Stromal-Cell-Derived Factor 1 on Stem-Cell Homing and Tissue Regeneration in Ischaemic Cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Abbott, J.D.; Huang, Y.; Liu, D.; Hickey, R.; Krause, D.S.; Giordano, F.J. Stromal Cell-Derived Factor-1alpha Plays a Critical Role in Stem Cell Recruitment to the Heart after Myocardial Infarction but Is Not Sufficient to Induce Homing in the Absence of Injury. Circulation 2004, 110, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Li, J.; Liao, L.; Chen, B.; Li, B.; Chen, L.; Jia, H.; Zhao, R.C. Regulation of CXCR4 Expression in Human Mesenchymal Stem Cells by Cytokine Treatment: Role in Homing Efficiency in NOD/SCID Mice. Haematologica 2007, 92, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, Q.; Yan, J.; Hu, R.; Jiang, H. Isoflurane Preconditioning Promotes the Survival and Migration of Bone Marrow Stromal Cells. CPB 2015, 36, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Zhang, N.; Wang, H.-W.; Gao, P.; Yang, Q.-P.; Wen, Q.-P. CXCR4 Receptor Overexpression in Mesenchymal Stem Cells Facilitates Treatment of Acute Lung Injury in Rats. J. Biol. Chem. 2015, 290, 1994–2006. [Google Scholar] [CrossRef] [Green Version]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the Tumour Stroma to Improve Cancer Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of Immune Cells and Cytokines on Inflammation and Immunosuppression in the Tumor Microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef] [PubMed]
- Bertzbach, L.D.; Ip, W.-H.; Dobner, T. Animal Models in Human Adenovirus Research. Biology 2021, 10, 1253. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilson, R.; Krutzke, L.; Wienen, F.; Rojewski, M.; Zeplin, P.H.; Funk, W.; Schrezenmeier, H.; Kochanek, S.; Kritzinger, A. Evaluation of Human Mesenchymal Stromal Cells as Carriers for the Delivery of Oncolytic HAdV-5 to Head and Neck Squamous Cell Carcinomas. Viruses 2023, 15, 218. https://doi.org/10.3390/v15010218
Nilson R, Krutzke L, Wienen F, Rojewski M, Zeplin PH, Funk W, Schrezenmeier H, Kochanek S, Kritzinger A. Evaluation of Human Mesenchymal Stromal Cells as Carriers for the Delivery of Oncolytic HAdV-5 to Head and Neck Squamous Cell Carcinomas. Viruses. 2023; 15(1):218. https://doi.org/10.3390/v15010218
Chicago/Turabian StyleNilson, Robin, Lea Krutzke, Frederik Wienen, Markus Rojewski, Philip Helge Zeplin, Wolfgang Funk, Hubert Schrezenmeier, Stefan Kochanek, and Astrid Kritzinger. 2023. "Evaluation of Human Mesenchymal Stromal Cells as Carriers for the Delivery of Oncolytic HAdV-5 to Head and Neck Squamous Cell Carcinomas" Viruses 15, no. 1: 218. https://doi.org/10.3390/v15010218
APA StyleNilson, R., Krutzke, L., Wienen, F., Rojewski, M., Zeplin, P. H., Funk, W., Schrezenmeier, H., Kochanek, S., & Kritzinger, A. (2023). Evaluation of Human Mesenchymal Stromal Cells as Carriers for the Delivery of Oncolytic HAdV-5 to Head and Neck Squamous Cell Carcinomas. Viruses, 15(1), 218. https://doi.org/10.3390/v15010218